




Guarda gli articoli recenti
Abstract
Introduzione: La malattia di Kikuchi-Fujimoto, o linfoadenite istiocitica necrotizzante, è una condizione rara con un decorso benigno che imita la malignità alla presentazione. Un esame accurato e la conferma istologica della diagnosi sono quindi fondamentali.
Presentazione del caso: Una donna di 53 anni che si è presentata con febbre, linfoadenopatia generalizzata e sintomi costituzionali è stata sottoposta a biopsia linfonodale dopo aver temuto un linfoma maligno.
Gestione ed esito: Al paziente è stata diagnosticata la malattia di Kikuchi-Fujimoto dopo la conferma istopatologica della necrosi con abbondanti istiociti e neutrofili assenti.
Discussione: La febbre e la linfoadenopatia hanno un’ampia differenziazione, e una mancata diagnosi di una condizione rara come la malattia di Kikuchi-Fujimoto può portare a un trattamento inappropriato in una condizione altrimenti benigna.
Introduzione
La malattia di Kikuchi-Fujimoto (KFD), o linfoadenite istiocitica necrotizzante di Kikuchi, è una condizione benigna e autolimitata caratterizzata tipicamente da febbre e linfoadenopatia focale. La KFD ha una distribuzione mondiale e colpisce entrambi i sessi con una maggiore prevalenza nelle giovani donne e nelle persone asiatiche. La vera incidenza non è nota. L’eziologia della KFD è ancora dibattuta in letteratura, tuttavia sono stati implicati agenti infettivi e studi più recenti sono suggestivi di un’eziologia virale basata sull’istologia e la clinicopatologia associate. Anche se un’associazione con il lupus eritematoso sistemico (SLE) è stata discussa in tutta la letteratura, nessuna relazione definitiva è stata dimostrata. È interessante notare che il LES deve ancora essere escluso prima di confermare una diagnosi. La KFD può presentarsi con sintomi che tipicamente durano da uno a quattro mesi, ma la recidiva
è stata riportata nel 3-4% degli individui affetti. Presentiamo un caso di KFD in una donna con febbre, linfoadenopatia generalizzata e sintomi sistemici che sono stati diagnosticati dalla biopsia linfonodale, che è stata inizialmente ordinata sulla base di un sospetto di linfoma maligno.
Presentazione del caso
Una donna afroamericana di 53 anni si è presentata con una storia di due settimane di febbre, sudorazione notturna, affaticamento, perdita di peso involontaria e linfoadenopatia dolente nel collo. Ha ammesso una storia di sei mesi di linfoadenopatia altrettanto dolorosa nell’ascella. La sua storia era significativa per le emicranie così come il dolore cronico alla schiena auto-medicato con la marijuana. Uno screening dell’antigene HIV di un anno prima era negativo.
L’esame fisico ha rivelato linfonodi teneri e palpabili nella catena cervicale posteriore, nell’area auricolare, nell’ascella e nell’area femorale. Il linfonodo più grande è stato trovato nella catena cervicale posteriore destra e misurava 1,43 cm all’ecografia (Figura 1).

Figura 1. Linfonodo di 1,43 cm nella catena cervicale posteriore destra all’ecografia
I risultati di laboratorio iniziali hanno rivelato un conteggio dei globuli bianchi (WBC) di 3.000 composto per il 50% da linfociti, una velocità di eritrosedimentazione (ESR) di 105, e proteina C-reattiva (CRP) di 32,3. La VES e la CRP ottenute l’anno precedente erano rispettivamente 10 e 1,6. ANA, lupus anticoagulante e anticorpi anti-Smith erano tutti negativi. Una puntura lombare era notevole per il CSF con un WBC di 12 e proteine di 47. L’antigene EBV e le IgG erano positivi, così come le IgG della varicella. Le colture del CSF e lo striscio acido-resistente erano negativi, così come le colture dei tessuti, del sangue e dei funghi. Anche lo screening della sifilide, la PCR del CMV e la sierologia per l’antigene del criptococco, i coccidioidi e la toxoplasmosi erano negativi. La TAC di contrasto del collo e del torace ha dimostrato un nodulo polmonare di 5 mm, nessun linfonodo ingrossato e nessuna prova di embolia polmonare acuta.
I sintomi costituzionali del paziente erano inizialmente preoccupanti per una nuova diagnosi di linfoma maligno, ed è stata eseguita una biopsia del nucleo guidata dall’ecografia e un FNA di un linfonodo nel triangolo cervicale posteriore destro. La biopsia ha dimostrato sezioni di linfonodo con aree focali di necrosi all’interno della corteccia caratterizzata da materiale fibrinoide contenente numerosi piccoli frammenti nucleari (Figura 2). All’interno e intorno a questi focolai c’era una popolazione di istiociti e cellule linfoidi trasformate. Le cellule di plasma erano focalmente presenti, ma i neutrofili e l’infiammazione granulomatosa erano assenti (Figura 3). La colorazione immunoistochimica ha rivelato istiociti CD68 positivi con pochi linfociti B CD20 positivi nelle aree necrotiche e molte cellule più grandi CD3 positive coerenti con l’origine delle cellule T.
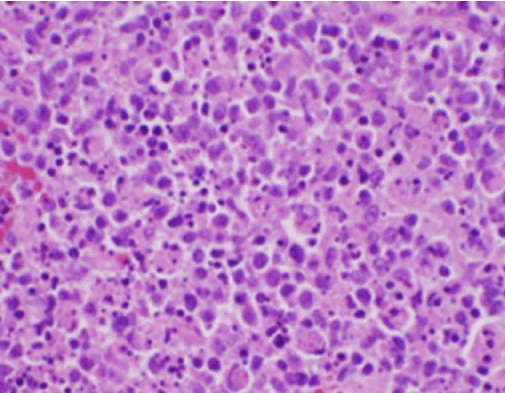
Figura 2. Colorazione H&E della biopsia del linfonodo che dimostra necrosi e neutrofili assenti
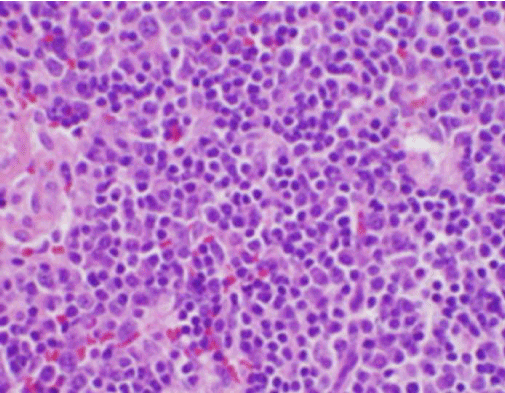
Figura 3. H&Macchia E della biopsia del linfonodo cervicale che mostra numerosi istiociti e neutrofili assenti
La paziente è stata diagnosticata con KFD sulla base dei risultati istogologici di linfoadenite necrotizzante, istiociti CD68 positivi, necrosi in assenza di neutrofili e un’abbondanza di istiociti. Il resto del suo decorso ospedaliero non è stato movimentato ed è stata successivamente dimessa con una terapia a base di glucocorticoidi e un follow-up in reumatologia.
Discussione
La malattia di Kikuchi-Fujimoto, o linfoadenite istiocitica necrotizzante, è stata descritta per la prima volta in giovani donne in Giappone nel 1972 e da allora è stata oggetto di studio in tutta la letteratura. È caratterizzata da febbre e linfoadenopatia, ma può essere presente una varietà di segni e sintomi associati, tra cui sintomi sistemici come affaticamento, perdita di peso, sudorazione notturna, e altre manifestazioni meno comuni come meningite asettica, splenomegalia, eruzione cutanea e artralgie. La KFD si presenta più comunemente come febbre e linfoadenopatia cervicale unilaterale nelle giovani donne, tuttavia la linfoadenopatia può anche essere multifocale. Il coinvolgimento extranodale della pelle e del midollo osseo, per esempio, è spesso accompagnato da sintomi sistemici, tra cui sudorazione notturna, perdita di peso, affaticamento, nausea, vomito e diarrea.
La febbre e la linfoadenopatia osservate nella KFD comportano una diagnosi differenziale che è abbastanza ampia e comprende eziologie infettive, autoimmuni e maligne. La diagnosi differenziale per la febbre e la linfoadenopatia è diversa e comprende un’ampia gamma di malattie con diverse gamme di prognosi. Diagnosticare erroneamente un paziente con una condizione che ha una prognosi sfavorevole e, in particolare, un regime di trattamento più aggressivo può portare a cure inappropriate e a conseguenze mediche negative. Molteplici rapporti hanno dimostrato che la sintomatologia della KFD è stata confusa con condizioni come la tubercolosi e il linfoma maligno. Come tale, è importante per i medici essere consapevoli e includere la malattia di Kikuchi-Fujimoto, anche se una condizione rara con una presentazione variabile, all’interno della differenziale per la febbre e la linfoadenopatia.
L’esatta patogenesi rimane incerta, tuttavia clinicamente può assomigliare a una serie di condizioni che portano prognosi più gravi, come la tubercolosi (TB), toxoplasmosi, sifilide, linfogranuloma venereo, SLE e linfoma. In una serie di casi di Fujimoto, il 58% dei pazienti è stato erroneamente diagnosticato e trattato in modo inappropriato per la TB linfonodale prima che l’istopatologia confermasse la diagnosi di KFD. Un rapporto ha discusso un caso di KFD con manifestazioni cutanee che è stato inizialmente diagnosticato come un linfoma a grandi cellule. Il paziente è stato sottoposto a un ciclo di terapia citotossica prima che venisse fatta la diagnosi corretta.
La diagnosi di KFD viene fatta sulla biopsia linfonodale che dimostra foci paracorticali di necrosi coagulativa con detriti carioretici e un infiltrato cellulare istiocitico. Il reperto istopatologico di detriti nucleari frammentati suggerisce l’apoptosi come meccanismo di morte cellulare, e i risultati immunoistochimici, compreso l’aumento delle cellule T Fas/FasL e CD 8+, supportano ulteriormente questo meccanismo. I risultati di laboratorio sono in genere normali, ma possono verificarsi una VES elevata e leucopenia. Inoltre, i risultati di laboratorio possono mostrare leucopenia, VES elevata e anemia. Non esiste un trattamento efficace al di là della gestione dei sintomi con antipiretici, analgesici
e riposo. Tuttavia alcuni rapporti hanno dimostrato che i pazienti con sintomi gravi possono beneficiare di corticosteroidi.
Conclusione
La malattia di Kikuchi-Fujimoto è rara, ma la sua conoscenza da parte dei medici è fondamentale quando si considera la diagnosi differenziale per un paziente che presenta sintomi di febbre e linfoadenopatia. La KFD è una condizione auto-limitata con una prognosi eccellente e la gestione è tipicamente di supporto con riposo e FANS per alleviare i sintomi. Quando viene scambiata per una malattia più grave, come la tubercolosi o il linfoma maligno, i pazienti possono essere inutilmente sottoposti a trattamenti aggressivi.
- Bosch X, Guilabert A, Miquel R, Campo E (2004) Enigmatica malattia di Kikuchi-Fujimoto: una revisione completa. Am J Clin Pathol 122: 141-152.
- Chiu CF, Chow KC, Lin TY, Tsai MH, Shih CM, et al. (2000) Infezione da virus in pazienti con linfoadenite istiocitica necrotizzante a Taiwan. Rilevamento del virus di Epstein-Barr, del virus linfotropo delle cellule T umane di tipo I e del parvovirus B19. Am J Clin Pathol 113: 774-781.
- Tsang WY, Chan JK, Ng CS (1994) Linfadenite di Kikuchi. Un’analisi morfologica di 75 casi con particolare riferimento alle caratteristiche insolite. Am J Surg Pathol 18: 219-231.
- Dorfman RF, Berry GJ (1988) Linfadenite istiocitica necrotizzante di Kikuchi: un’analisi di 108 casi con enfasi sulla diagnosi differenziale. Semin Diagn Pathol 5: 329-345.
- Aneja A, Maheswari K U1, H J GD1, Sheikh S2 (2014) Un raro caso di linfoadenopatia multifocale in un giovane maschio. Oxf Med Case Reports 2014: 141-144.