




Bekijk de Recente artikelen
Abstract
Inleiding: De ziekte van Kikuchi-Fujimoto, of histiocytaire necrotiserende lymfadenitis, is een zeldzame aandoening met een goedaardig beloop die bij presentatie een maligniteit nabootst. Zorgvuldig onderzoek en histologische bevestiging van de diagnose is daarom van cruciaal belang.
Presentatie van het geval: Een 53-jarige vrouw die zich presenteerde met koorts, gegeneraliseerde lymfadenopathie en constitutionele symptomen onderging een lymfeklierbiopsie na bezorgdheid voor maligne lymfomen.
Behandeling en uitkomst: De patiënt werd gediagnosticeerd met de Ziekte van Kikuchi-Fujimoto na histopathologische bevestiging van necrose met overvloedige histiocyten en afwezige neutrofielen.
Discussie: Koorts en lymfadenopathie dragen een breed differentieel met zich mee, en een gemiste diagnose van een zeldzame aandoening als de ziekte van Kikuchi-Fujimoto kan leiden tot een ongepaste behandeling bij een anders goedaardige aandoening.
Inleiding
De ziekte van Kikuchi-Fujimoto (KFD), of Kikuchi histiocytaire necrotiserende lymfadenitis, is een goedaardige, zelfbegrensde aandoening die typisch wordt gekenmerkt door koorts en focale lymfadenopathie. KFD is wereldwijd verspreid en treft beide geslachten, met een hogere prevalentie bij jonge vrouwen en Aziaten. De werkelijke incidentie is niet bekend. De etiologie van KFD wordt nog steeds besproken in de literatuur, maar infectieuze agentia zijn geïmpliceerd, en meer recente studies suggereren een virale etiologie gebaseerd op de geassocieerde histologie en klinische pathologie. Hoewel een verband met systemische lupus erythematosous (SLE) in de literatuur ter discussie staat, is er geen definitief verband aangetoond. Interessant is dat SLE nog steeds moet worden uitgesloten alvorens de diagnose te bevestigen. KFD kan zich presenteren met symptomen die typisch één tot vier maanden duren, maar recidief
is gerapporteerd in 3-4% van de getroffen personen. Wij presenteren een geval van KFD bij een vrouw met koorts, gegeneraliseerde lymfadenopathie en systemische symptomen die werden gediagnosticeerd door een lymfeklierbiopsie, die aanvankelijk werd besteld op basis van een verdenking op maligne lymfoom.
Casepresentatie
Een 53-jarige Afro-Amerikaanse vrouw presenteerde zich met een twee weken durende geschiedenis van koorts, nachtelijk zweten, vermoeidheid, onbedoeld gewichtsverlies en pijnlijke lymfadenopathie in de nek. Ze gaf een zes maanden lange geschiedenis van een soortgelijke pijnlijke lymfadenopathie in de oksel toe. In haar verleden had ze veel hoofdpijn en chronische rugpijn, waarvoor ze zelf marihuana gebruikte. Een HIV-antigeenscreening van een jaar eerder was negatief.
Lichamelijk onderzoek toonde gevoelige, palpabele lymfeklieren in de achterste halsketen, de oorschelpstreek, de axilla en het dijbeengebied. De grootste knoop werd gevonden in de rechter achterste cervicale keten en mat 1,43 cm bij echografie (Figuur 1).

Figuur 1. 1,43 cm lymfeklier in de rechter achterste halsketen op echografie
Initiële laboratoriumresultaten toonden een aantal witte bloedcellen (WBC) van 3000 bestaande uit 50% lymfocyten, een erytrocytenbezinkingssnelheid (ESR) van 105, en C-reactief proteïne (CRP) van 32,3. De ESR en CRP van het voorafgaande jaar bedroegen respectievelijk 10 en 1,6. ANA, lupus anticoagulans, en anti-smith antilichamen waren allemaal negatief. Een lumbaalpunctie was opmerkelijk voor CSF met een WBC van 12 en proteïne van 47. EBV antigeen en IgG was positief, evenals Varicella IgG. CSF kweken en zuurvaste uitstrijkjes waren negatief, evenals weefsel-, bloed- en schimmelkweken. Syfilis screening, CMV PCR, en serologie voor cryptokokken-antigen, coccidioïden, en toxoplasmose waren ook negatief. Contrast CT van de hals en borst toonde een 5 mm pulmonale nodule, geen vergrote lymfeklieren, en geen aanwijzingen voor acute longembolie.
De constitutionele symptomen van de patiënt waren aanvankelijk zorgwekkend voor een nieuwe diagnose van maligne lymfoom, en een echogeleide kernbiopsie en FNA van een lymfeklier in de rechter achterste cervicale driehoek werd uitgevoerd. Biopsie toonde secties van de lymfeklier met focale gebieden van necrose binnen de cortex gekenmerkt door fibrinoid materiaal met talrijke kleine nucleaire fragmenten (Figuur 2). Binnen en rondom deze foci was een populatie van histiocyten en getransformeerde lymfoïde cellen. Plasmacellen waren focaal aanwezig, maar neutrofielen en granulomateuze ontsteking waren afwezig (figuur 3). Immunohistochemische kleuring toonde CD68-positieve histiocyten met enkele CD20-positieve B-lymfocyten in de necrotische gebieden en veel grotere, CD3-positieve cellen die consistent waren met T-celoorsprong.
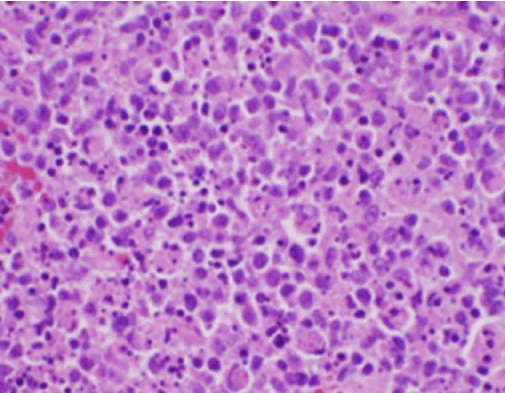
Figuur 2. H&E-kleuring van lymfeklierbiopsie met necrose en afwezige neutrofielen
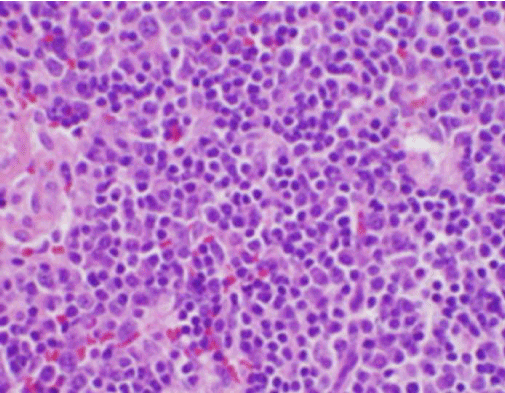
Figuur 3. H&E-kleuring van cervicale lymfeklierbiopsie toont talrijke histiocyten en afwezige neutrofielen
De diagnose KFD werd gesteld op basis van de histogologische bevindingen van necrotiserende lymfadenitis, CD68 positieve histiocyten, necrose in afwezigheid van neutrofielen, en een overvloed aan histiocyten. De rest van het ziekenhuisverloop verliep zonder problemen en zij werd ontslagen op glucocorticoïdtherapie met follow-up door reumatologie.
Discussie
De ziekte van Kikuchi-Fujimoto, of histiocytaire necrotiserende lymfadenitis, werd voor het eerst beschreven bij jonge vrouwen in Japan in 1972 en is sindsdien een onderwerp van studie in de literatuur. De ziekte wordt gekenmerkt door koorts en lymfadenopathie, maar er kunnen verschillende geassocieerde tekens en symptomen aanwezig zijn, waaronder systemische symptomen zoals vermoeidheid, gewichtsverlies, nachtelijk zweten, en andere minder vaak voorkomende manifestaties zoals aseptische meningitis, splenomegalie, huiduitslag en artralgieën. KFD presenteert zich meestal als koorts en unilaterale cervicale lymfadenopathie bij jonge vrouwen, hoewel de lymfadenopathie ook multifocaal kan zijn. Extranodale betrokkenheid van de huid en het beenmerg, bijvoorbeeld, gaat vaak gepaard met systemische symptomen, waaronder zweten ’s nachts, gewichtsverlies, vermoeidheid, misselijkheid, braken en diarree.
De koorts en lymfadenopathie die bij KFD worden gezien, dragen een differentiële diagnose met zich mee die vrij breed is en infectieuze, auto-immune, en maligne etiologieën omvat. De differentiële diagnose voor koorts en lymfadenopathie is divers en omvat een breed scala van ziekten met verschillende prognoses. Het foutief diagnosticeren van een patiënt met een aandoening met een ongunstige prognose en, in het bijzonder, een agressiever behandelingsschema kan leiden tot ongepaste zorg en slechte medische gevolgen. Meerdere rapporten hebben aangetoond dat de symptomatologie van KFD wordt verward met aandoeningen zoals tuberculose en maligne lymfoom. Als zodanig is het belangrijk voor clinici om zich bewust te zijn van de ziekte van Kikuchi-Fujimoto en deze op te nemen in het differentieel voor koorts en lymfadenopathie, ook al is het een zeldzame aandoening met een variabele presentatie.
De exacte pathogenese blijft onzeker, maar het kan klinisch lijken op een aantal aandoeningen met een meer ernstige prognose, zoals tuberculose (TB), toxoplasmose, syfilis, lymphogranuloma venereum, SLE, en lymfoom. In een case serie door Fujimoto werd 58% van de patiënten verkeerd gediagnosticeerd en ongeschikt behandeld voor lymfekliertbc voordat histopathologie de diagnose van KFD bevestigde. Eén verslag besprak een geval van KFD met cutane manifestaties dat aanvankelijk werd gediagnosticeerd als een grootcellig lymfoom. De patiënt onderging één kuur cytotoxische therapie voordat de juiste diagnose werd gesteld.
De diagnose van KFD wordt gesteld op basis van een lymfeklierbiopsie die paracorticale foci van coagulatieve necrose met karyorrhectisch debris en een histiocytair cellulair infiltraat aantoont. De histopathologische bevinding van gefragmenteerd nucleair debris suggereert apoptose als het mechanisme van celdood, en immunohistochemische bevindingen, waaronder verhoogde Fas/FasL en CD 8 + T-cellen, ondersteunt verder dit mechanisme . De laboratoriumresultaten zijn doorgaans normaal, maar een verhoogde ESR en leukopenie kunnen voorkomen. Daarnaast kunnen de laboratoriumresultaten leukopenie, verhoogde ESR en anemie laten zien. Er is geen effectieve behandeling buiten symptoombestrijding met antipyretica, analgetica,
en rust. Sommige rapporten hebben echter aangetoond dat patiënten met ernstige symptomen baat kunnen hebben bij corticosteroïden.
Conclusie
De ziekte van Kikuchi-Fujimoto is zeldzaam, maar bekendheid ermee bij clinici is van cruciaal belang bij het overwegen van de differentiële diagnose voor een patiënt die zich presenteert met symptomen van koorts en lymfadenopathie. KFD is een op zichzelf staande aandoening met een uitstekende prognose, en de behandeling is meestal ondersteunend met rust en NSAID’s om de symptomen te verlichten. Wanneer het wordt aangezien voor een ernstiger ziekte, zoals TB of maligne lymfoom, kunnen patiënten onnodig worden onderworpen aan agressieve behandelingen.
- Bosch X, Guilabert A, Miquel R, Campo E (2004) Enigmatische ziekte van Kikuchi-Fujimoto: een uitgebreid overzicht. Am J Clin Pathol 122: 141-152.
- Chiu CF, Chow KC, Lin TY, Tsai MH, Shih CM, et al. (2000) Virusinfectie bij patiënten met histiocytic necrotizing lymphadenitis in Taiwan. Detectie van Epstein-Barr virus, type I humaan T-cel lymfotroop virus, en parvovirus B19. Am J Clin Pathol 113: 774-781.
- Tsang WY, Chan JK, Ng CS (1994) Kikuchi’s lymphadenitis. A morphologic analysis of 75 cases with special reference to unusual features. Am J Surg Pathol 18: 219-231.
- Dorfman RF, Berry GJ (1988) Kikuchi’s histiocytic necrotizing lymphadenitis: an analysis of 108 cases with emphasis on differential diagnosis. Semin Diagn Pathol 5: 329-345.
- Aneja A, Maheswari K U1, H J GD1, Sheikh S2 (2014) Een zeldzaam geval van multifocale lymfadenopathie bij een jonge man. Oxf Med Case Reports 2014: 141-144.