




Se de seneste artikler
Abstract
Introduktion: Kikuchi-Fujimoto-sygdom, eller histiocytisk nekrotiserende lymfadenitis, er en sjælden tilstand med et benignt forløb, der efterligner malignitet ved præsentationen. En omhyggelig undersøgelse og histologisk bekræftelse af diagnosen er derfor afgørende.
Præsentation af tilfælde: En 53-årig kvinde, der præsenterede sig med feber, generaliseret lymfadenopati og konstitutionelle symptomer, blev underkastet lymfeknudebiopsi efter bekymring for malignt lymfom.
Behandling og resultat: Patienten blev diagnosticeret med Kikuchi-Fujimoto-sygdom efter histopatologisk bekræftelse af nekrose med rigelige histiocytter og fraværende neutrofile.
Diskussion:
Indledning
Kikuchi-Fujimoto Disease (KFD), eller Kikuchi histiocytisk nekrotiserende lymfadenitis, er en godartet, selvbegrænsende tilstand, der typisk er karakteriseret ved feber og fokal lymfadenopati. KFD er udbredt over hele verden og rammer begge køn med en højere prævalens hos unge kvinder og asiatiske folk . Den reelle forekomst er ikke kendt. Ætiologien til KFD er stadig omdiskuteret i litteraturen, men infektiøse agenser har været impliceret, og nyere undersøgelser tyder på en viral ætiologi på baggrund af den tilknyttede histologi og klinisk patologi . Selv om en sammenhæng med systemisk lupus erythematosous (SLE) er blevet diskuteret i hele litteraturen, er der ikke blevet påvist nogen endelig sammenhæng. Det er interessant, at SLE stadig skal udelukkes, før diagnosen kan bekræftes. KFD kan præsentere med symptomer, der typisk varer en til fire måneder, men recidiv
er blevet rapporteret hos 3-4 % af de berørte personer . Vi præsenterer et tilfælde af KFD hos en kvinde med feber, generaliseret lymfadenopati og systemiske symptomer, der blev diagnosticeret ved lymfeknudebiopsi, som oprindeligt blev bestilt på baggrund af en mistanke om malignt lymfom.
Faldpræsentation
En 53-årig afroamerikansk kvinde præsenterede sig med en to ugers historie med feber, nattesved, træthed, utilsigtet vægttab og smertefuld lymfadenopati i nakken. Hun indrømmede en seks måneders historie med tilsvarende smertefuld lymfadenopati i axilla. Hendes anamnese var signifikant for hovedpine samt kroniske rygsmerter, som hun selvmedicinerede med marihuana. En HIV-antigen-screening fra et år tidligere var negativ.
Fysisk undersøgelse afslørede ømme, palpable lymfeknuder i den bageste cervikale kæde, aurikulært område, axilla og femoralt område. Den største knude blev fundet i den højre bageste halskæde og målte 1,43 cm ved ultralyd (figur 1).

Figur 1. 1,43 cm lymfeknude i højre bageste halskæde på ultralyd
Initiale laboratorieresultater afslørede et antal hvide blodlegemer (WBC) på 3.000 bestående af 50% lymfocytter, en erytrocytsedimentationshastighed (ESR) på 105 og C-reaktivt protein (CRP) på 32,3. Baseline ESR og CRP fra det foregående år var henholdsvis 10 og 1,6. ANA, lupus antikoagulerende antistoffer og anti-smith-antistoffer var alle negative. En lumbalpunktur var bemærkelsesværdig for CSF med en WBC på 12 og et protein på 47. EBV-antigen og IgG var positiv samt Varicella IgG. CSF-kulturer og syrefaste smear var negative, ligesom vævs-, blod- og svampekulturer var negative. Syfilis-screening, CMV-PCR og serologi for kryptokok-antigen, coccidioider og toxoplasmose var også negative. Kontrast CT af hals og bryst viste en 5 mm lungeknude, ingen forstørrede lymfeknuder og ingen tegn på akut lungeemboli.
Patientens konstitutionelle symptomer var i første omgang bekymrende for en ny diagnose af malignt lymfom, og der blev foretaget en ultralydsvejledt kernebiopsi og FNA af en lymfeknude i højre bageste cervikale trekant. Biopsien viste sektioner af lymfeknuden med fokale områder af nekrose i cortexen karakteriseret ved fibrinoid materiale indeholdende talrige små kernefragmenter (Figur 2). Inden for og omkring disse foci var der en population af histiocytter og transformerede lymfoide celler. Plasmaceller var fokuseret til stede, men neutrofile og granulomatøs inflammation var fraværende (figur 3). Immunhistokemisk farvning afslørede CD68-positive histiocytter med få CD20-positive B-lymfocytter i de nekrotiske områder og mange større, CD3-positive celler, der var i overensstemmelse med T-celleoprindelse.
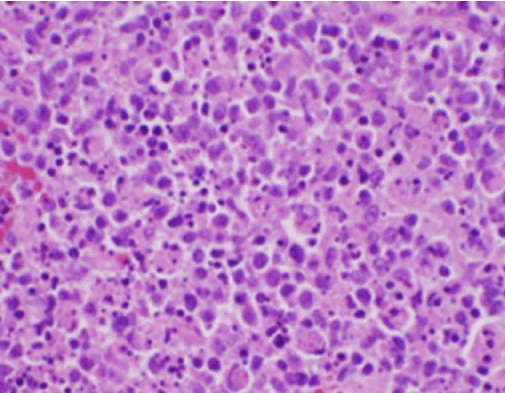
Figur 2. H&E-farve af lymfeknudebiopsi, der viser nekrose og fravær af neutrofiler
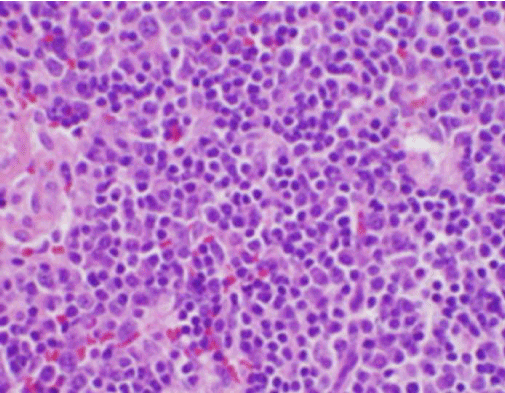
Figur 3. H&E farvning af biopsi af cervikal lymfeknude, der viser talrige histiocytter og fraværende neutrofiler
Patienten blev diagnosticeret med KFD på baggrund af de histogologiske fund af nekrotiserende lymfadenitis, CD68-positive histiocytter, nekrose i fravær af neutrofiler og en overflod af histiocytter. Resten af hendes hospitalsforløb var begivenhedsløst, og hun blev efterfølgende udskrevet med glukokortikoidbehandling med opfølgning hos reumatologien.
Diskussion
Kikuchi-Fujimoto-sygdom, eller histiocytær nekrotiserende lymfadenitis, blev første gang beskrevet hos unge kvinder i Japan i 1972 og har siden været genstand for undersøgelser i hele litteraturen. Den er karakteriseret ved feber og lymfadenopati, men en række associerede tegn og symptomer kan være til stede, herunder systemiske symptomer som træthed, vægttab, nattesved, og andre mindre almindelige manifestationer som aseptisk meningitis, splenomegali, udslæt og arthralgias . KFD viser sig oftest som feber og unilateral cervikal lymfadenopati hos unge kvinder, men lymfadenopatien kan også være multifokal. Extranodal involvering af hud og knoglemarv, for eksempel, er ofte ledsaget af systemiske symptomer, herunder nattesved, vægttab, træthed, kvalme, opkastning og diarré .
Den feber og lymfadenopati, der ses i KFD, bærer en differentialdiagnose, der er ret bred og omfatter infektiøse, autoimmune og maligne ætiologier. Differentialdiagnosen for feber og lymfadenopati er forskelligartet og omfatter en bred vifte af sygdomme med varierende prognoser. Fejldiagnosticering af en patient med en tilstand, der har en ugunstig prognose, og især et mere aggressivt behandlingsregime kan føre til uhensigtsmæssig behandling og dårlige medicinske konsekvenser. Flere rapporter har vist, at symptomatologien ved KFD er blevet forvekslet med tilstande som tuberkulose og malignt lymfom . Som sådan er det vigtigt for klinikere at være opmærksomme på og inkludere Kikuchi-Fujimoto-sygdom, om end en sjælden tilstand med en variabel præsentation, i differentialdiagnosen for feber og lymfadenopati.
Den nøjagtige patogenese er fortsat usikker, men den kan klinisk ligne en række tilstande, der bærer mere alvorlige prognoser, såsom tuberkulose (TB), toxoplasmose, syfilis, lymfogranuloma venereum, SLE og lymfom . I en sagsserie af Fujimoto blev 58 % af patienterne fejldiagnosticeret og uhensigtsmæssigt behandlet for lymfeknude-tbc, før histopatologien bekræftede diagnosen KFD . En rapport omtalte et tilfælde af KFD med kutane manifestationer, der oprindeligt blev diagnosticeret som et storcellet lymfom . Patienten gennemgik et forløb med cytotoksisk terapi, før den korrekte diagnose blev stillet .
Diagnosen KFD stilles ved lymfeknudebiopsi, der viser parakortikale foci af koagulativ nekrose med karyorrhectic debris og et histiocytisk cellulært infiltrat . Det histopatologiske fund af fragmenterede kerneaffald tyder på apoptose som mekanisme for celledød, og immunhistokemiske fund, herunder øget Fas/FasL og CD 8+ T-celler, understøtter yderligere denne mekanisme . Laboratoriefundene er typisk normale, men forhøjet ESR og leukopeni kan forekomme. Desuden kan laboratoriefundene vise leukopeni, forhøjet ESR og anæmi. Der findes ingen effektiv behandling ud over symptombehandling med febernedsættende midler, analgetika,
og hvile. Nogle rapporter har dog vist, at patienter med alvorlige symptomer kan have gavn af kortikosteroider.
Konklusion
Kikuchi-Fujimoto-sygdom er sjælden, men kendskabet til den blandt klinikere er afgørende, når man overvejer differentialdiagnosen for en patient, der præsenterer sig med symptomer på feber og lymfadenopati. KFD er en selvbegrænsende tilstand med en fremragende prognose, og behandlingen er typisk understøttende med hvile og NSAID’er til at lindre symptomerne. Når den forveksles med en mere alvorlig sygdom, såsom tuberkulose eller malignt lymfom, kan patienterne unødigt udsættes for aggressiv behandling.
- Bosch X, Guilabert A, Miquel R, Campo E (2004) Enigmatic Kikuchi-Fujimoto disease: a comprehensive review. Am J Clin Pathol 122: 141-152.
- Chiu CF, Chow KC, Lin TY, Tsai MH, Shih CM, et al. (2000) Virusinfektion hos patienter med histiocytær nekrotiserende lymfadenitis i Taiwan. Påvisning af Epstein-Barr-virus, human T-cell lymphotropisk virus af type I og parvovirus B19. Am J Clin Pathol 113: 774-781.
- Tsang WY, Chan JK, Ng CS (1994) Kikuchi’s lymphadenitis. En morfologisk analyse af 75 tilfælde med særlig henvisning til usædvanlige træk. Am J Surg Pathol 18: 219-231.
- Dorfman RF, Berry GJ (1988) Kikuchi’s histiocytic necrotizing lymphadenitis: an analysis of 108 cases with emphasis on differential diagnosis. Semin Diagn Pathol 5: 329-345.
- Aneja A, Maheswari K U1, H J GD1, H J GD1, Sheikh S2 (2014) A rare case of multifocal lymphadenopathy in a young male. Oxf Med Case Reports 2014: 141-144.