




Mecanismo – Cómo el ibuprofeno (Advil; Motrin) inhibe la actividad antitrombótica de la aspirina
Cualquier persona que realice una simple búsqueda bibliográfica se encontrará con una plétora de artículos que discuten, debaten e incluso discuten sobre este tema.Las razones de todas estas cuestiones, en nuestra opinión, tienen que ver con las numerosas inconsistencias entre los estudios en cuanto a las dosis y la duración del ibuprofeno utilizado, el momento de la dosificación del ibuprofeno en relación con la administración de aspirina, la dosis y la formulación (recubrimiento entérico o no entérico) de la aspirina utilizada, la población de pacientes estudiada (voluntarios sanos frente a pacientes con enfermedades cardiovasculares (ECV) conocidas), si se utilizaron marcadores de laboratorio sustitutivos en lugar de pruebas que realmente evalúan la agregación plaquetaria y, por último, el diseño del estudio utilizado por los investigadores para obtener sus resultados1-6. Por lo tanto, es muy difícil, o casi imposible, extrapolar los datos actuales de cada uno de estos estudios, todos los cuales tienen limitaciones o inconsistencias entre sí, y generar una respuesta definitiva que pueda traducirse en puntos finales clínicamente significativos que sean aplicables a la población general. Algunas de las discrepancias en la bibliografía pueden deberse a la capacidad de las plaquetas para agregarse en momentos en los que las concentraciones de los fármacos antiinflamatorios no esteroideos (AINE) son bajas, en comparación con los primeros momentos después de la administración, cuando las concentraciones son mayores.7 Cuando el ibuprofeno se libera del sitio de unión en la COX-1, parte de la aspirina ya se habrá eliminado del organismo.
La Administración de Alimentos y Medicamentos (FDA) advirtió recientemente a los profesionales de la salud: «Los pacientes que utilicen aspirina de liberación inmediata (no con recubrimiento entérico) y tomen una dosis única de ibuprofeno de 400 mg deben tomar el ibuprofeno al menos 30 minutos o más después de la ingestión de aspirina, o más de 8 horas antes de la ingestión de aspirina para evitar la atenuación del efecto de ésta. Además, hay varios estudios con resultados contradictorios».8 Basándose en esta recomendación, el propósito de este número no es criticar cada uno de los estudios publicados sobre este tema, sino más bien explicar el mecanismo propuesto para la interacción de los fármacos y, a continuación, destacar algunos de los problemas con su interpretación en relación con la literatura médica.
¿Qué ocurre durante la agregación plaquetaria normal?
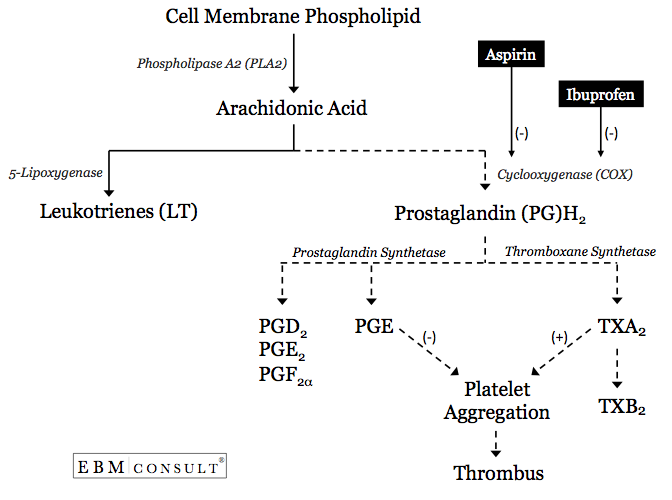
- El proceso de aumento de la agregación plaquetaria comienza con la liberación de ácido araquidónico (AA) de la membrana celular de la plaqueta.6
- El AA puede seguir una de las dos vías metabólicas, la vía de la lipoxigenasa, que generará leucotrienos (LT), o la vía de la ciclooxigenasa (COX)-1 para formar prostaglandina (PG) H2.9 La prostaglandina H2 puede ser metabolizada por la prostaglandinsintetasa para formar otros PG o puede ser metabolizada por la tromboxano (TX)sintetasa para formar TXA2.
- Si se forma TXA2 se facilitará o fomentará la agregación plaquetaria.10 Esto ocurre específicamente cuando el AA es capaz de viajar a través de un canal hidrofóbico donde puede hacer contacto con el sitio catalítico dentro de la enzima COX-1. Si este canal o las áreas que rodean el sitio catalítico están bloqueados, el AA no podrá ser metabolizado en PGH2 y luego en TXA2, reduciendo así la probabilidad de agregación plaquetaria.6,9,11
¿Cómo interfiere entonces la aspirina en la agregación plaquetaria?
- Al administrar la aspirina, ésta acetila irreversiblemente un residuo de serina en la posición 529 dentro del canal hidrofóbico, que se encuentra muy cerca del sitio catalítico donde el AA puede ser metabolizado en TXA2 derivado de las plaquetas por la enzima COX-1.6,9,11 La acetilación de esta zona crea un bloqueo en el que el AA no podrá acceder al sitio catalítico dentro de la COX-1.
- Como la aspirina hace esto de forma irreversible, la capacidad de ese sitio catalítico dentro de la enzima COX-1 para metabolizar el AA queda bloqueada o inhibida durante toda la vida de esa plaqueta (normalmente unos 7-12 días). Esta es una de las principales razones por las que la aspirina confiere un beneficio cardioprotector contra los eventos cardiovasculares cuando se utiliza principalmente para la prevención secundaria.
- Por lo tanto, si algo más compite o bloquea el acceso de la aspirina a acetilar este residuo de serina dentro de la enzima COX-1, los beneficios cardioprotectores pueden disminuir.
¿Cómo interfiere el ibuprofeno en la actividad farmacológica de la aspirina?
- Al igual que todos los AINE, el ibuprofeno es un inhibidor reversible y competitivo del sitio catalítico para el metabolismo del AA dentro del canal hidrofóbico de la enzima COX-1.7,12
- La presencia de ibuprofeno en este canal hidrofóbico bloquea de forma competitiva el acceso de la aspirina para acetilar el residuo de serina que se encuentra cerca del sitio catalítico para el AA.7,12,13
- El grado de inhibición del acceso de la aspirina para ejercer su efecto farmacológico por parte del ibuprofeno va a estar influenciado por una serie de factores.
El primer factor, y el más obvio, tiene que ver con el orden en que se administran la aspirina y el ibuprofeno entre sí. Si la aspirina se administra primero, accederá a acetilar irreversiblemente el residuo de serina dentro de la enzima COX-1. Recuerde, una vez que la aspirina ha inhibido irreversiblemente la enzima COX-1, el efecto antiplaquetario seguirá existiendo de por vida para esa plaqueta. El siguiente factor es la concentración de ibuprofeno presente en relación con el tiempo de coadministración de la aspirina. Dado que la inhibición del ibuprofeno es competitiva, la agregación plaquetaria no sólo está influida por la concentración de ibuprofeno presente, sino que también es de naturaleza reversible. Por lo tanto, a medida que los niveles del fármaco disminuyen a través de las vías de eliminación, la cantidad de ibuprofeno capaz de bloquear el acceso de la aspirina a su sitio activo también disminuye, especialmente teniendo en cuenta su corta vida media de 2 a 4 horas.14 Esta característica farmacocinética del ibuprofeno es la razón por la que debe volver a dosificarse varias veces a lo largo del día, mientras que la aspirina sólo se dosifica una vez al día. Por lo tanto, se puede ver por qué hay variaciones en los resultados de múltiples estudios publicados en la literatura médica. Así pues, el impacto clínico de esta interacción farmacológica está influenciado por el orden de administración de los dos medicamentos, la dosis y la formulación de aspirina utilizada, la dosis y la frecuencia de administración de ibuprofeno utilizada, la población de pacientes estudiada y el tipo de criterio de valoración de ese estudio.
Al final, la verdadera cuestión es si esta interacción se traduce en un resultado cardiovascular predefinido y clínicamente relevante orientado al paciente. Hasta donde sabemos, no se ha realizado ningún ensayo clínico prospectivo de diseño adecuado para responder a esta pregunta con datos convincentes en la población de pacientes en cuestión.
- GengoFM, Rubin L, Robson M et al. Effects of ibuprofen on the magnitude andduration of aspirin’s inhibition of platelet aggregation: clinical consequencesin stroke prophylaxis. J Clin Pharmacol 2008;48:117-22.
- GladdingPA, Webster MWI, Farrell HB et al. The antiplatelet effect of sixnon-steroidal anti-inflammatory drugs and their pharmacodynamic interactionswith aspirin in healthy volunteers. Am J Cardiol 2008;101:1060-1063.
- CryerB, Berlin RG, Copper SA et al. Double-blind, randomized, parallel,placebo-controlled study of ibuprofen effects on thromboxane B2 concentrationsin aspirin-treated healthy adult volunteers. Clin Ther2005;27:185-191.
- MacDonaldTM, Wei L et al. Effect of ibuprofen on cardioprotective effect ofaspirin. Lancet 2003;361:573-74.
- KurthT, Glynn RJ, Walker AM et al. Inhibition of clinical benefits of aspirinon first myocardial infarction by nonsteroidal antiinflammatory drugs. Circulation 2003;108:1191-1195.
- Catella-LawsonF, Reilly MP, Kapoor SC et al. Cyclooxygenase inhibitors and theantiplatelet effects of aspirin. N Engl J Med 2001;345:1809-17.
- EvansAM. Pharmacodynamics and pharmacokinetics of the profens:enantioselectivity, clinical implications, and special reference toS(+)-ibuprofen. J Clin Pharmacol 1996;36:7S-15S.
- Food& Drug Administration. Información para profesionales sanitarios:uso concomitante de ibuprofeno y aspirina. Departamento de Salud de EE.UU. &Servicios Humanos. Último acceso: 09-19-2011.
- FunkCD, Funk LB, Kennedy ME et al. Human platelet/erythroleukemia cellprostaglandin G/H synthase: cDNA cloning, expression, and gene chromosomalassignment. FASEB J 1991;5:2304-12.
- FitzgeraldGA. Mechanisms of platelet activation: thromboxane A2 as an amplifyingsignal for other agonists. Am J Cardiol 1991;68:11B-15B.
- LollPJ, Picot D, Garavito RM. The structural basis of aspirin activityinferred from the crystal structure of inactivated prostaglandin H2synthase. Nat Struct Biol 1995;2:637-43.
- LollPJ, Picot D, Ekabo O et al. Synthesis and use of iodinated nonsteroidalantiinflammatory drug analogs as crystallographic probes of the prostaglandinH2 synthase cyclooxygenase active site. Biochemistry 1996;35:7330-40.
- RaoGH, Johnson GG, Reddy KR et al. Ibuprofen protects plateletcyclooxygenase from irreversible inhibition by aspirin. Arteriosclerosis 1983;3:383-8.