




Mira los artículos recientes
Abstract
Introducción: La enfermedad de Kikuchi-Fujimoto, o linfadenitis necrotizante histiocítica, es una afección rara con un curso benigno que imita una neoplasia en su presentación. Por lo tanto, el examen cuidadoso y la confirmación histológica del diagnóstico son fundamentales.
Presentación del caso: Una mujer de 53 años que se presentó con fiebre, linfadenopatía generalizada y síntomas constitucionales fue sometida a una biopsia de los ganglios linfáticos tras la preocupación por un linfoma maligno.
Manejo y resultado: El paciente fue diagnosticado de enfermedad de Kikuchi-Fujimoto tras la confirmación histopatológica de necrosis con abundantes histiocitos y ausencia de neutrófilos.
Discusión: La fiebre y la linfadenopatía conllevan un amplio diferencial, y un diagnóstico erróneo de una afección rara como la enfermedad de Kikuchi-Fujimoto puede conducir a un tratamiento inadecuado en una afección que de otro modo sería benigna.
Introducción
La enfermedad de Kikuchi-Fujimoto (KFD), o linfadenitis necrotizante histiocítica de Kikuchi, es una afección benigna y autolimitada que se caracteriza típicamente por fiebre y linfadenopatía focal. La KFD se distribuye por todo el mundo y afecta a ambos sexos, con una mayor prevalencia en las mujeres jóvenes y los asiáticos. No se conoce la verdadera incidencia. La etiología de la KFD sigue siendo objeto de debate en la literatura, aunque se han implicado agentes infecciosos, y los estudios más recientes sugieren una etiología viral basada en la histología y la clinicopatología asociadas. Aunque se ha debatido en la literatura una asociación con el lupus eritematoso sistémico (LES), no se ha demostrado una relación definitiva. Curiosamente, el LES debe descartarse antes de confirmar el diagnóstico. La KFD puede presentarse con síntomas que suelen durar de uno a cuatro meses, aunque se ha informado de la recurrencia
en el 3-4% de los individuos afectados. Presentamos un caso de KFD en una mujer con fiebre, linfadenopatía generalizada y síntomas sistémicos que se diagnosticaron mediante una biopsia de los ganglios linfáticos, que se ordenó inicialmente basándose en la sospecha de un linfoma maligno.
Presentación del caso
Una mujer afroamericana de 53 años se presentó con una historia de dos semanas de fiebre, sudores nocturnos, fatiga, pérdida de peso involuntaria y linfadenopatía dolorosa en el cuello. Admitió una historia de seis meses de linfadenopatía igualmente dolorosa en la axila. Sus antecedentes eran significativos por cefaleas, así como por dolor de espalda crónico automedicado con marihuana. Una prueba de antígeno del VIH de un año antes fue negativa.
El examen físico reveló ganglios linfáticos sensibles y palpables en la cadena cervical posterior, la zona auricular, la axila y la zona femoral. El ganglio más grande se encontraba en la cadena cervical posterior derecha y medía 1,43 cm por ecografía (Figura 1).

Figura 1. Ganglio linfático de 1,43 cm en la cadena cervical posterior derecha en la ecografía
Los resultados de laboratorio iniciales revelaron un recuento de glóbulos blancos (WBC) de 3.000 compuesto por un 50% de linfocitos, una velocidad de sedimentación globular (ESR) de 105 y una proteína C reactiva (CRP) de 32,3. La VSG y la PCR de referencia obtenidas el año anterior eran 10 y 1,6, respectivamente. Los ANA, el anticoagulante lúpico y los anticuerpos anti-smith fueron todos negativos. En la punción lumbar se observó un LCR con un recuento de leucocitos de 12 y proteínas de 47. El antígeno e IgG del VEB fue positivo, así como la IgG de la varicela. Los cultivos de LCR y el frotis ácido-resistente fueron negativos, así como los cultivos de tejidos, sangre y hongos. El cribado de sífilis, la PCR para CMV y la serología para antígeno criptocócico, coccidioides y toxoplasmosis también fueron negativos. La tomografía computarizada con contraste del cuello y el tórax demostró un nódulo pulmonar de 5 mm, sin agrandamiento de los ganglios linfáticos, y sin evidencia de embolia pulmonar aguda.
Los síntomas constitucionales de la paciente fueron inicialmente preocupantes para un nuevo diagnóstico de linfoma maligno, y se realizó una biopsia central guiada por ultrasonido y FNA de un ganglio linfático en el triángulo cervical posterior derecho. La biopsia mostró secciones del ganglio linfático con áreas focales de necrosis dentro de la corteza caracterizadas por material fibrinoide que contenía numerosos fragmentos nucleares pequeños (Figura 2). Dentro y alrededor de estos focos había una población de histiocitos y células linfoides transformadas. Las células plasmáticas estaban presentes focalmente, pero los neutrófilos y la inflamación granulomatosa estaban ausentes (Figura 3). La tinción inmunohistoquímica reveló histiocitos CD68 positivos con pocos linfocitos B CD20 positivos en las áreas necróticas y muchas células más grandes, CD3 positivas, consistentes con el origen de las células T.
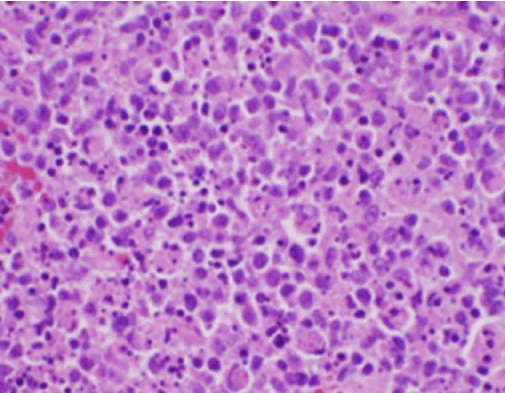
Figura 2. Tinción H&E de biopsia de ganglio linfático que demuestra necrosis y ausencia de neutrófilos
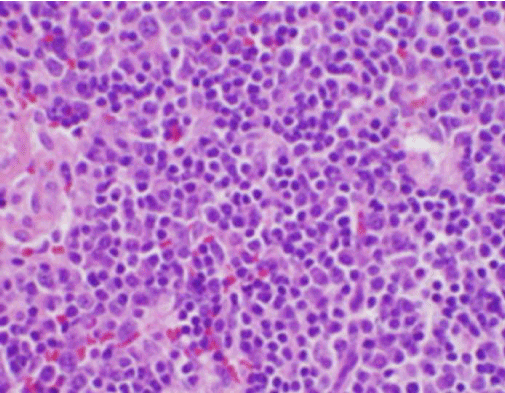
Figura 3. Tinción H&E de biopsia de ganglio linfático cervical que muestra numerosos histiocitos y ausencia de neutrófilos
La paciente fue diagnosticada de KFD en base a los hallazgos histogológicos de linfadenitis necrotizante, histiocitos CD68 positivos, necrosis en ausencia de neutrófilos y abundancia de histiocitos. El resto de su evolución hospitalaria fue tranquila y posteriormente fue dada de alta con tratamiento de glucocorticoides y seguimiento por reumatología.
Discusión
La enfermedad de Kikuchi-Fujimoto, o linfadenitis necrotizante histiocítica, fue descrita por primera vez en mujeres jóvenes en Japón en 1972 y desde entonces ha sido objeto de estudio en toda la literatura. Se caracteriza por fiebre y linfadenopatía, aunque puede haber una variedad de signos y síntomas asociados, incluyendo síntomas sistémicos como fatiga, pérdida de peso, sudores nocturnos y otras manifestaciones menos comunes como meningitis aséptica, esplenomegalia, erupción cutánea y artralgias . La enfermedad de KFD se presenta más comúnmente como fiebre y linfadenopatía cervical unilateral en mujeres jóvenes, aunque la linfadenopatía también puede ser multifocal. La afectación extraganglionar de la piel y la médula ósea, por ejemplo, suele ir acompañada de síntomas sistémicos, como sudores nocturnos, pérdida de peso, fatiga, náuseas, vómitos y diarrea.
La fiebre y la linfadenopatía que se observan en la enfermedad de KFD conllevan un diagnóstico diferencial bastante amplio que incluye etiologías infecciosas, autoinmunes y malignas. El diagnóstico diferencial de la fiebre y la linfadenopatía es diverso y abarca una gran variedad de enfermedades con distintos pronósticos. Diagnosticar erróneamente a un paciente con una enfermedad que tiene un pronóstico desfavorable y, en particular, un régimen de tratamiento más agresivo puede conducir a una atención inadecuada y a malas consecuencias médicas. Múltiples informes han demostrado que la sintomatología del KFD se ha confundido con afecciones como la tuberculosis y el linfoma maligno . Por lo tanto, es importante que los médicos conozcan e incluyan la enfermedad de Kikuchi-Fujimoto, aunque es una afección rara con una presentación variable, dentro del diferencial para la fiebre y la linfadenopatía.
La patogénesis exacta sigue siendo incierta, sin embargo, puede parecerse clínicamente a una serie de afecciones que conllevan un pronóstico más grave, como la tuberculosis (TB), la toxoplasmosis, la sífilis, el linfogranuloma venéreo, el LES y el linfoma . En una serie de casos de Fujimoto, el 58% de los pacientes fueron diagnosticados erróneamente y tratados de forma inapropiada por tuberculosis en los ganglios linfáticos antes de que la histopatología confirmara el diagnóstico de DFK. En un informe se describe un caso de KFD con manifestaciones cutáneas que se diagnosticó inicialmente como un linfoma de células grandes. El paciente fue sometido a un curso de terapia citotóxica antes de que se hiciera el diagnóstico correcto.
El diagnóstico de KFD se hace en la biopsia de los ganglios linfáticos que demuestra focos paracorticales de necrosis coagulativa con restos carióticos y un infiltrado celular histiocítico . El hallazgo histopatológico de restos nucleares fragmentados sugiere que la apoptosis es el mecanismo de muerte celular, y los hallazgos inmunohistoquímicos, incluyendo el aumento de Fas/FasL y de células T CD 8+, apoyan aún más este mecanismo. Los hallazgos de laboratorio suelen ser normales, pero puede producirse una elevación de la VSG y leucopenia. Además, los resultados de laboratorio pueden mostrar leucopenia, elevación de la VSG y anemia. No existe un tratamiento eficaz más allá del manejo de los síntomas con antipiréticos, analgésicos,
y reposo. Sin embargo, algunos informes han demostrado que los pacientes con síntomas graves pueden beneficiarse de los corticosteroides.
Conclusión
La enfermedad de Kikuchi-Fujimoto es poco frecuente, pero su conocimiento por parte de los médicos es crucial a la hora de considerar el diagnóstico diferencial de un paciente que presenta síntomas de fiebre y linfadenopatía. La enfermedad de KFD es una afección autolimitada con un pronóstico excelente, y el tratamiento suele ser de apoyo con reposo y AINE para aliviar los síntomas. Cuando se confunde con una enfermedad más grave, como la tuberculosis o el linfoma maligno, los pacientes pueden ser sometidos innecesariamente a tratamientos agresivos.
- Bosch X, Guilabert A, Miquel R, Campo E (2004) Enigmatic Kikuchi-Fujimoto disease: a comprehensive review. Am J Clin Pathol 122: 141-152.
- Chiu CF, Chow KC, Lin TY, Tsai MH, Shih CM, et al. (2000) Virus infection in patients with histiocytic necrotizing lymphadenitis in Taiwan. Detección del virus de Epstein-Barr, del virus linfotrópico de células T humanas de tipo I y del parvovirus B19. Am J Clin Pathol 113: 774-781.
- Tsang WY, Chan JK, Ng CS (1994) Kikuchi’s lymphadenitis. Un análisis morfológico de 75 casos con especial referencia a las características inusuales. Am J Surg Pathol 18: 219-231.
- Dorfman RF, Berry GJ (1988) Kikuchi’s histiocytic necrotizing lymphadenitis: an analysis of 108 cases with emphasis on differential diagnosis. Semin Diagn Pathol 5: 329-345.
- Aneja A, Maheswari K U1, H J GD1, Sheikh S2 (2014) Un caso raro de linfadenopatía multifocal en un hombre joven. Oxf Med Case Reports 2014: 141-144.