




Le blog du génotypage


. 

Rosario García
Il y a quelque temps, nous avons parlé des trisomies qui se produisent chez les humains, et nous avons fait remarquer qu’il existe un groupe distinct de trisomies qui sont exceptionnellement compatibles avec la vie. Ces trisomies sont celles causées par la duplication ou la double hérédité de l’un des chromosomes sexuels, X et Y.
Les trisomies qui se produisent dans les chromosomes sexuels peuvent passer inaperçues pendant longtemps. Parfois, les personnes qui en sont atteintes l’ont découvert parce qu’elles ont obtenu un résultat positif à un test génétique qu’elles passaient dans un autre but.
Le fait que la plupart de ces trisomies passent inaperçues est dû au fait qu’en présence de plus d’un chromosome X, l’un d’entre eux est sélectionné « au hasard » et les autres sont inactivés, chacun d’entre eux formant un corpuscule de Barr, qui est simplement une « boule » d’ADN provenant du chromosome X inactivé. L’inactivation du chromosome X a lieu pour empêcher la surexpression des gènes sur le chromosome lorsqu’ils ne sont nécessaires qu’en dose unique. Après cette inactivation, seule l’expression des gènes sur le chromosome X sélectionné se produit, ce qui entraîne un phénotype normal ou quasi-normal.
Regardons certaines de ces trisomies qui peuvent se produire sur les chromosomes sexuels :
Syndrome du triple X (XXX)
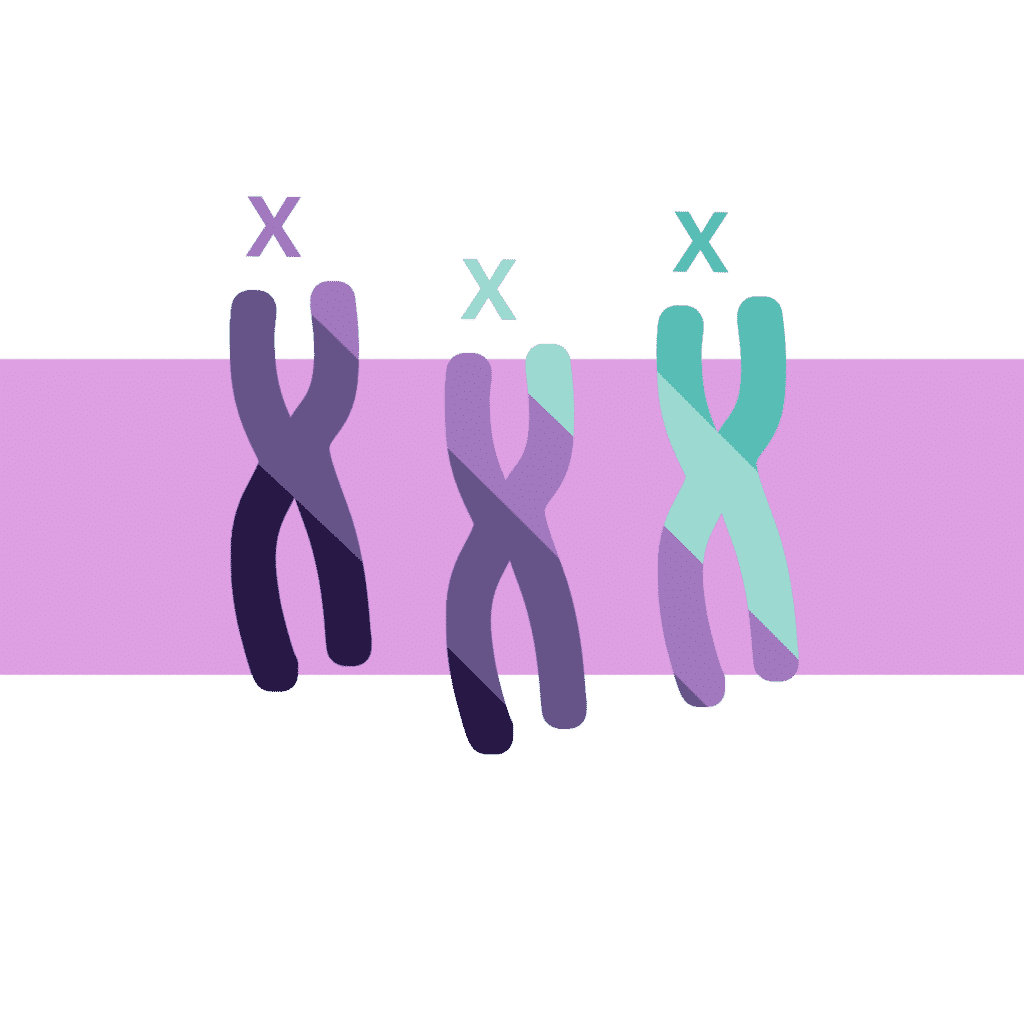
L’incidence du syndrome du triple X est d’un cas pour 1 000 filles nées vivantes, bien que cela puisse n’être qu’une estimation, car la plupart des personnes atteintes du syndrome atteignent l’âge adulte sans le savoir et sans être diagnostiquées.
Le syndrome du triple X implique l’héritage d’un chromosome X supplémentaire, de sorte qu’il y a 47 chromosomes dans le génome au lieu des 46 habituels. La présence de ce chromosome X supplémentaire est due à des échecs sporadiques dans la formation des gamètes chez les parents. Il convient également de mentionner que le risque de ces erreurs dans la formation des ovules et des spermatozoïdes augmente avec l’âge du père et de la mère. On a également constaté que, dans le cas du syndrome du triple X, la plupart de ces erreurs se produisent dans les ovules.
Bien que la plupart des chromosomes X supplémentaires proviennent de ces erreurs dans la formation des gamètes, on a observé qu’environ 20 % des chromosomes X supplémentaires proviennent d’une duplication d’un des deux chromosomes X dans les premiers stades de la division du zygote. En outre, ce type de duplication au début du développement est le type de cause le plus courant de la présence du chromosome X supplémentaire en mosaïque, ce qui se produit chez les personnes qui ont le chromosome supplémentaire dans certaines de leurs cellules et pas dans d’autres.
Concernant les symptômes qui apparaissent dans ce syndrome, on pense qu’ils sont causés par la surexpression de certains gènes du chromosome X qui échappent à l’inactivation. Normalement, ces gènes non activés dans le corpuscule de Barr contribuent à un phénotype normal puisqu’ils sont nécessaires à « double dose ». Mais dans le syndrome du triple X, ils présentent une dose d’expression plus élevée : « triple dose », qui peut aller jusqu’au quadruple ou au quintuple (dans le cas des tétra- et pentasomies du chromosome X, qui existent aussi et se produisent parfois). Rappelez-vous que pour chaque chromosome X supplémentaire, il y aura un corpuscule de Barr de plus.
Les symptômes ne sont pas toujours communs à tous les individus atteints et sont très divers. Les symptômes les plus courants sont les suivants : augmentation de la taille, difficultés d’apprentissage (notamment en matière de motricité et de langage), hypotonie, problèmes rénaux et parfois crises d’épilepsie.
Les autres symptômes qui ont été rapportés en relation avec le syndrome du triple X sont les suivants : plis épicanthaux (comme un pli dans la paupière qui recouvre légèrement la zone interne de l’œil où se trouve le canal lacrymal), courbure anormale de l’auriculaire, microcéphalie, certains problèmes de coordination, risque accru de scoliose, faible estime de soi et risque accru d’anxiété et/ou de dépression, QI légèrement inférieur à la moyenne, dyslexie, troubles du langage, yeux anormalement écarquillés, TDAH (trouble déficitaire de l’attention avec hyperactivité), absence ou malformation des ovaires et/ou de l’utérus, perte de la fonction ovarienne bien avant la ménopause, retard de puberté ou puberté précoce, pieds plats, infections urinaires, etc.
Malgré tous les symptômes susmentionnés, les personnes touchées par ce syndrome ne présentent généralement pas beaucoup de traits physiques caractéristiques, et n’ont généralement pas de problèmes de fertilité ou de reproduction. En outre, un grand nombre de personnes touchées sont asymptomatiques (ne présentent aucun signe indiquant qu’elles ont le syndrome), ce qui contribue au fait qu’elles peuvent ne pas être conscientes qu’elles ont le chromosome X supplémentaire
Le traitement des patients atteints du syndrome du triple X consiste généralement en une aide psychologique et éducative pour compenser les retards d’apprentissage, des conseils sur les traitements de fertilité et le soutien des organisations de personnes touchées. Cependant, comme mentionné ci-dessus, les symptômes peuvent être très légers et il est très facile pour les personnes concernées de mener une vie normale.
Syndrome de Klinefelter (XXY)
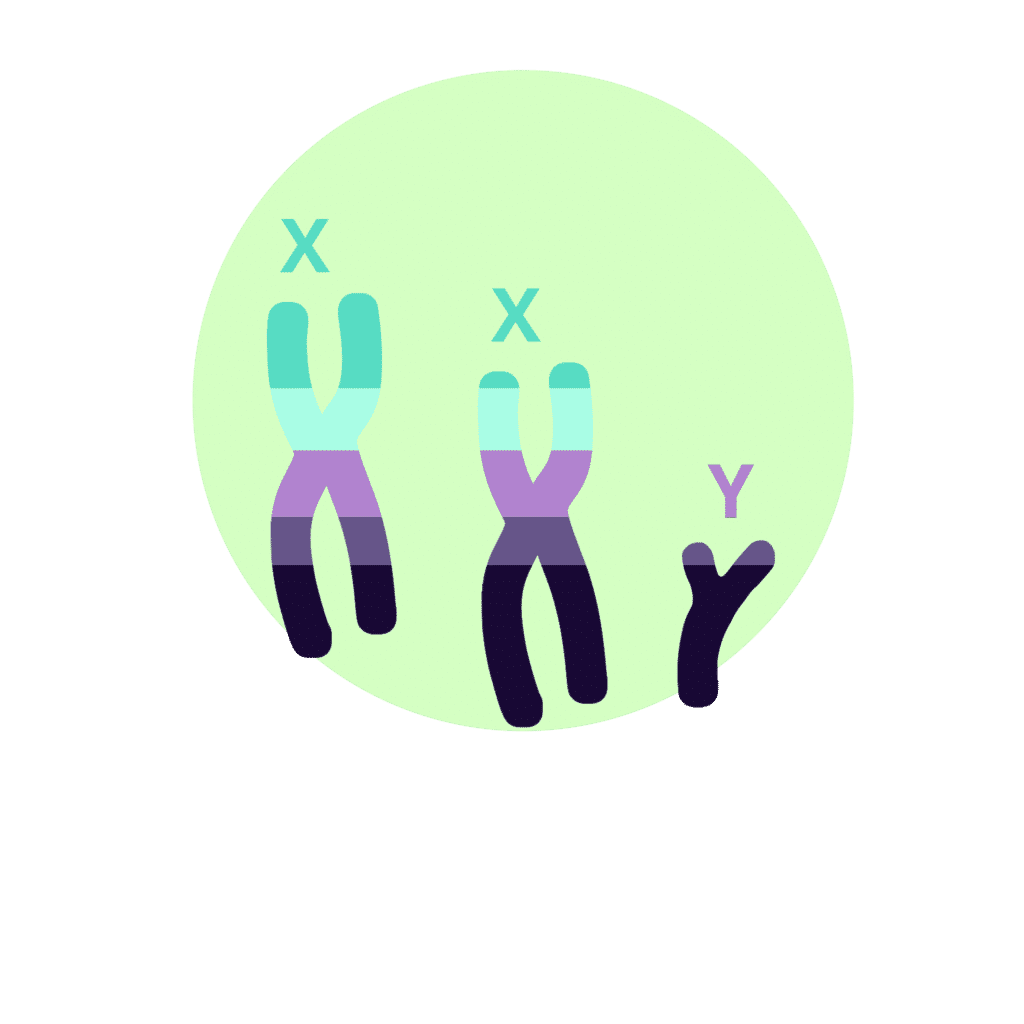
Le syndrome de Klinefelter a une incidence d’environ 1 atteint pour 500 naissances vivantes. Cependant, comme pour le syndrome du triple X, il est possible que ce chiffre ne soit qu’une estimation, car une grande partie des personnes touchées par le syndrome de Klinefelter atteignent l’âge adulte sans savoir qu’elles en sont atteintes.
Ce syndrome survient chez les personnes qui possèdent une paire de chromosomes XY et entre un et trois chromosomes X supplémentaires (XXY, XXXY, XXXXY). Par conséquent, ces personnes ont respectivement 47, 48 ou 49 chromosomes dans leurs cellules. Le plus souvent, un seul chromosome X supplémentaire est présent, et les symptômes sont d’autant plus graves que le nombre de chromosomes X supplémentaires dans les cellules de l’individu est élevé. D’autre part, un corpuscule de Barr apparaît également pour chaque chromosome X supplémentaire dans la paire XY.
Dans le cas du syndrome de Klinefelter, la duplication (ou triplication, ou quadruplication) du chromosome X se produit plus fréquemment lors des premières divisions du zygote, ce qui est dû à une erreur dans la formation des gamètes des parents (ovules XX ou spermatozoïdes XY, selon le parent chez qui l’erreur se produit). Bien que la cause la plus fréquente du syndrome soit des erreurs au cours du développement précoce du zygote, cela n’implique pas qu’il ne puisse pas également être causé par un échec dans la formation des gamètes.
De nombreux symptômes qui peuvent se produire chez les personnes touchées par le syndrome de Klinefelter ont été décrits, car il a été l’objet d’études pendant de nombreuses années, et donc de nombreuses données et signes du syndrome ont été collectés.
Le principal symptôme est l’infertilité, dont la cause peut être due à une diminution du taux de testostérone ou de la fonction endocrinienne, à des testicules plus petits et parfois à une azoospermie (absence totale de spermatozoïdes et incapacité à en produire) chez les individus porteurs de chromosomes XXY.
Les autres symptômes qui peuvent également être caractéristiques sont : la gynécomastie ou l’augmentation du volume des seins (bien que cela ne soit généralement constaté que chez un tiers des personnes atteintes du syndrome de Klinefelter), l’augmentation de la taille, la diminution de la quantité et de la répartition des poils du visage et du corps, la diminution des capacités de coordination, un certain degré d’hypotonie et des hanches un peu plus larges.
Ces caractéristiques apparaissent à partir de la puberté et peuvent avoir des degrés d’implication variables. En fait, seulement 10 % environ des personnes atteintes du syndrome de Klinefelter envisagent de subir une opération d’ablation des seins, et beaucoup se font opérer pour réduire le risque de cancer du sein.
Peu de caractéristiques phénotypiques permettraient de différencier un individu XXY adulte d’un individu XY. En effet, de nombreuses personnes affectées par le syndrome XXY ne sont diagnostiquées que lorsqu’elles se rendent dans une clinique de fertilité ou de reproduction assistée.
Le syndrome de Klinefelter peut également être détecté par un diagnostic génétique prénatal ou diagnostiqué pendant la puberté, lorsque de légères altérations du développement des caractères sexuels secondaires sont présentes.
Comme indiqué ci-dessus, bien que le syndrome de Klinefelter n’ait pas de remède, certains des traitements visant à atténuer ou à réduire les symptômes comprennent la réduction mammaire, le traitement par la testostérone exogène ou l’hormonothérapie, les traitements de fertilité et de reproduction assistée, et le soutien éducatif et psychologique
Syndrome de XYYY

Le syndrome XYYY se produit chez environ 1 personne affectée pour 1 000 naissances vivantes. Parmi eux, seuls 30 % sont diagnostiqués avant la naissance et 20 % sont diagnostiqués à la puberté ou lorsque les troubles de l’apprentissage se manifestent.
Ce syndrome se caractérise par la présence d’un chromosome Y supplémentaire, de sorte que l’individu possède 47 chromosomes et que ses chromosomes sexuels sont XYY. La plupart des cas dans lesquels le chromosome Y supplémentaire est présent sont dus à des erreurs dans la formation des spermatozoïdes, de sorte que certains d’entre eux portent une charge génétique de YY au lieu de X ou Y. Et la fusion avec un ovule porteur du chromosome X produit le trio chromosomique XYY.
Comme dans les cas précédents, le syndrome XYY ne produit pas de symptômes trop marqués, et de nombreuses personnes atteintes ne savent pas qu’elles sont concernées. D’autre part, certains des symptômes les plus marquants peuvent inclure : une taille accrue (selon certaines études, environ 8 cm de plus que prévu), un risque accru d’acné, certains troubles de l’apprentissage et un retard dans le développement des compétences verbales.
Un tour de tête élargi, des dents anormalement grandes, des pieds plats, une augmentation de la graisse abdominale, une scoliose, des yeux anormalement écarquillés et une courbure de l’auriculaire vers les autres doigts ont également été rapportés comme signes physiques du syndrome XYY. Occasionnellement, des tics, des crises d’épilepsie, de l’asthme, un risque accru de TDAH, de dépression, d’anxiété et/ou de troubles du spectre autistique peuvent survenir.
Le syndrome XYY n’a pas de traitement spécifique, si ce n’est un soutien psychologique et éducatif pour les personnes touchées, car elles sont généralement asymptomatiques et peuvent mener une vie tout à fait normale. Comme mentionné ci-dessus, parce qu’elles ne présentent aucun symptôme, la plupart des personnes atteintes ne sont jamais diagnostiquées et ne savent jamais qu’elles ont un chromosome Y supplémentaire.
***
On a beaucoup spéculé sur certaines théories et faux mythes concernant les trisomies des chromosomes sexuels. Le terme super-femme n’est plus accepté pour les femmes atteintes du syndrome du triple X. Il n’est pas non plus vrai qu’un pourcentage plus élevé d’hommes atteints du syndrome de Klinelfelter sont homosexuels que la population masculine générale. Et rien ne prouve que les hommes atteints du syndrome XYY soient plus agressifs et plus enclins à commettre des infractions.
Toutes ces croyances découlent d’études plus anciennes qui intégraient les concepts sociaux de l’époque et ont maintenant été réfutées à plusieurs reprises. Les syndromes Triple X, Klinefelter et XYYY consistent uniquement en la présence d’un chromosome supplémentaire dans le génome d’une personne.
Vous voir dans un autre post !
Références
O. Robinson et P. A. Jacobs. L’origine du chromosome Y supplémentaire chez les hommes ayant un caryotype 47, XYYY. (1999). Génétique moléculaire humaine. Volume 8, numéro 12, pages 2205-2209.
.