




Regardez les articles récents
Abstract
Introduction : La maladie de Kikuchi-Fujimoto, ou lymphadénite nécrosante histiocytaire, est une affection rare d’évolution bénigne qui imite une tumeur maligne à la présentation. Un examen minutieux et la confirmation histologique du diagnostic sont donc essentiels.
Présentation du cas : Une femme de 53 ans qui a présenté de la fièvre, une lymphadénopathie généralisée et des symptômes constitutionnels a subi une biopsie des ganglions lymphatiques après avoir craint un lymphome malin.
Gestion et résultat : Le patient a reçu un diagnostic de maladie de Kikuchi-Fujimoto après confirmation histopathologique de la nécrose avec des histiocytes abondants et des neutrophiles absents.
Discussion : La fièvre et la lymphadénopathie portent un large différentiel, et un diagnostic manqué d’une condition rare comme la maladie de Kikuchi-Fujimoto peut conduire à un traitement inapproprié dans une condition autrement bénigne.
Introduction
La maladie de Kikuchi-Fujimoto (KFD), ou lymphadénite nécrosante histiocytaire de Kikuchi, est une condition bénigne, autolimitée, typiquement caractérisée par une fièvre et une lymphadénopathie focale. Le KFD est présent dans le monde entier et touche les deux sexes, avec une prévalence plus élevée chez les jeunes femmes et les Asiatiques. L’incidence réelle n’est pas connue. L’étiologie du KFD est encore débattue dans la littérature, mais des agents infectieux ont été impliqués et des études plus récentes suggèrent une étiologie virale sur la base de l’histologie et de la clinicopathologie associées. Bien qu’une association avec le lupus érythémateux systémique (LES) ait été débattue dans la littérature, aucune relation définitive n’a été prouvée. Il est intéressant de noter que le LED doit toujours être exclu avant de confirmer le diagnostic. La maladie de Creutzfeldt-Jakob peut se manifester par des symptômes qui durent généralement de un à quatre mois, mais une récurrence
a été signalée chez 3 à 4 % des personnes touchées. Nous présentons un cas de KFD chez une femme présentant une fièvre, une lymphadénopathie généralisée et des symptômes systémiques qui ont été diagnostiqués par une biopsie des ganglions lymphatiques, qui a été initialement ordonnée sur la base d’une suspicion de lymphome malin.
Présentation du cas
Une femme afro-américaine de 53 ans s’est présentée avec une histoire de deux semaines de fièvre, de sueurs nocturnes, de fatigue, de perte de poids involontaire et de lymphadénopathie douloureuse dans le cou. Elle a admis avoir souffert pendant six mois d’une lymphadénopathie douloureuse similaire au niveau de l’aisselle. Dans ses antécédents, elle présentait des maux de tête ainsi que des douleurs chroniques au dos, qu’elle soignait avec de la marijuana. Un dépistage de l’antigène du VIH effectué un an auparavant était négatif.
L’examen physique a révélé des ganglions lymphatiques sensibles et palpables dans la chaîne cervicale postérieure, la région auriculaire, l’aisselle et la région fémorale. Le plus gros ganglion a été trouvé dans la chaîne cervicale postérieure droite et mesurait 1,43 cm à l’échographie (figure 1).

Figure 1. Ganglion lymphatique de 1,43 cm dans la chaîne cervicale postérieure droite à l’échographie
Les résultats initiaux de laboratoire ont révélé une numération de globules blancs (WBC) de 3 000 composés de 50 % de lymphocytes, une vitesse de sédimentation érythrocytaire (ESR) de 105 et une protéine C-réactive (CRP) de 32,3. Les valeurs de base de l’ESR et de la CRP obtenues l’année précédente étaient respectivement de 10 et 1,6. Les anticorps ANA, lupus anticoagulant et anti-smith étaient tous négatifs. Une ponction lombaire a révélé un LCR avec un taux de globules blancs de 12 et de protéines de 47. L’antigène et les IgG de l’EBV étaient positifs ainsi que les IgG de la varicelle. Les cultures du LCR et le frottis acido-fast sont négatifs, ainsi que les cultures de tissus, de sang et de champignons. Le dépistage de la syphilis, la PCR du CMV et la sérologie pour l’antigène cryptococcique, les coccidioïdes et la toxoplasmose étaient également négatifs. La tomodensitométrie de contraste du cou et de la poitrine a montré un nodule pulmonaire de 5 mm, pas de ganglions lymphatiques hypertrophiés et aucun signe d’embolie pulmonaire aiguë.
Les symptômes constitutionnels du patient étaient initialement préoccupants pour un nouveau diagnostic de lymphome malin, et une biopsie à cœur échoguidée et une FNA d’un ganglion lymphatique dans le triangle cervical postérieur droit ont été réalisées. La biopsie a montré des sections de ganglion lymphatique avec des zones focales de nécrose dans le cortex caractérisées par un matériau fibrinoïde contenant de nombreux petits fragments nucléaires (Figure 2). À l’intérieur et autour de ces foyers se trouvait une population d’histiocytes et de cellules lymphoïdes transformées. Des plasmocytes étaient présents de façon focale, mais les neutrophiles et l’inflammation granulomateuse étaient absents (Figure 3). La coloration immunohistochimique a révélé des histiocytes positifs CD68 avec quelques lymphocytes B positifs CD20 dans les zones nécrotiques et de nombreuses cellules plus grandes, positives CD3, compatibles avec une origine de cellules T .
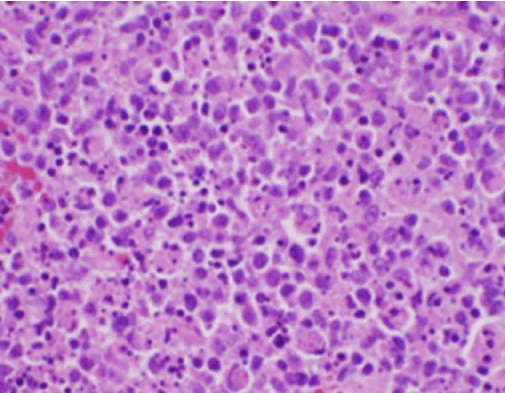
Figure 2. Coloration H&E d’une biopsie de ganglion lymphatique démontrant une nécrose et l’absence de neutrophiles
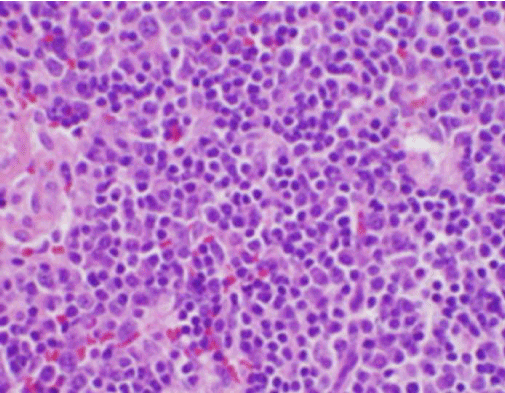
Figure 3. Coloration H&E d’une biopsie de ganglion cervical montrant de nombreux histiocytes et des neutrophiles absents
La patiente a été diagnostiquée avec une KFD sur la base des résultats histogologiques de la lymphadénite nécrosante, des histiocytes CD68 positifs, de la nécrose en l’absence de neutrophiles et d’une abondance d’histiocytes. Le reste de son parcours hospitalier s’est déroulé sans incident et elle est ensuite sortie de l’hôpital sous glucocorticothérapie avec un suivi en rhumatologie.
Discussion
La maladie de Kikuchi-Fujimoto, ou lymphadénite nécrosante histiocytaire, a été décrite pour la première fois chez des jeunes femmes au Japon en 1972 et a depuis été un sujet d’étude dans toute la littérature. Elle se caractérise par de la fièvre et une lymphadénopathie, mais divers signes et symptômes associés peuvent être présents, y compris des symptômes systémiques comme la fatigue, la perte de poids, les sueurs nocturnes, et d’autres manifestations moins courantes comme la méningite aseptique, la splénomégalie, l’éruption cutanée et les arthralgies . Le KFD se présente le plus souvent sous la forme d’une fièvre et d’une lymphadénopathie cervicale unilatérale chez les jeunes femmes, mais la lymphadénopathie peut aussi être multifocale. L’atteinte extranodale de la peau et de la moelle osseuse, par exemple, s’accompagne souvent de symptômes systémiques, notamment des sueurs nocturnes, une perte de poids, de la fatigue, des nausées, des vomissements et de la diarrhée .
La fièvre et la lymphadénopathie observées dans le KFD entraînent un diagnostic différentiel assez large qui inclut des étiologies infectieuses, auto-immunes et malignes. Le diagnostic différentiel de la fièvre et de la lymphadénopathie est diversifié et englobe un large éventail de maladies dont les pronostics varient. Poser un diagnostic erroné sur un patient atteint d’une maladie au pronostic défavorable et, en particulier, nécessitant un traitement plus agressif, peut conduire à des soins inappropriés et à de mauvaises conséquences médicales. De nombreux rapports ont démontré que la symptomatologie du KFD a été confondue avec des affections telles que la tuberculose et le lymphome malin. Il est donc important pour les cliniciens de connaître et d’inclure la maladie de Kikuchi-Fujimoto, bien qu’il s’agisse d’une condition rare avec une présentation variable, dans le différentiel pour la fièvre et la lymphadénopathie.
La pathogénie exacte reste incertaine, mais elle peut ressembler cliniquement à un certain nombre de conditions qui ont des pronostics plus graves, comme la tuberculose (TB), la toxoplasmose, la syphilis, le lymphogranulome vénérien, le LED et le lymphome . Dans une série de cas réalisée par Fujimoto, 58 % des patients ont été mal diagnostiqués et traités de manière inappropriée pour une tuberculose ganglionnaire avant que l’histopathologie ne confirme le diagnostic de KFD . Un rapport traite d’un cas de KFD avec des manifestations cutanées qui a été initialement diagnostiqué comme un lymphome à grandes cellules. Le patient a subi un traitement cytotoxique avant que le diagnostic correct ne soit posé.
Le diagnostic de KFD est posé sur une biopsie de ganglion lymphatique qui démontre des foyers paracorticaux de nécrose coagulante avec des débris caryorrhéiques et un infiltrat cellulaire histiocytaire. La découverte histopathologique de débris nucléaires fragmentés suggère que l’apoptose est le mécanisme de mort cellulaire, et les résultats immunohistochimiques, y compris l’augmentation de Fas/FasL et des cellules T CD 8+, confirment ce mécanisme. Les résultats de laboratoire sont généralement normaux, mais une élévation de la vitesse de sédimentation et une leucopénie peuvent se produire. De plus, les résultats de laboratoire peuvent montrer une leucopénie, un ESR élevé et une anémie. Il n’y a pas de traitement efficace au-delà de la gestion des symptômes avec des antipyrétiques, des analgésiques,
et du repos. Cependant, certains rapports ont montré que les patients présentant des symptômes sévères peuvent bénéficier de corticostéroïdes .
Conclusion
La maladie de Kikuchi-Fujimoto est rare, mais sa connaissance par les cliniciens est cruciale lors de l’examen du diagnostic différentiel pour un patient présentant des symptômes de fièvre et de lymphadénopathie. La maladie de Kikuchi-Fujimoto est une affection autolimitée avec un excellent pronostic, et la prise en charge est généralement de soutien avec du repos et des AINS pour soulager les symptômes. Lorsqu’elle est confondue avec une maladie plus grave, comme la tuberculose ou un lymphome malin, les patients peuvent être inutilement soumis à des traitements agressifs.
- Bosch X, Guilabert A, Miquel R, Campo E (2004) Enigmatic Kikuchi-Fujimoto disease : a comprehensive review. Am J Clin Catholic 122 : 141-152.
- Chiu CF, Chow KC, Lin TY, Tsai MH, Shih CM, et al. (2000) Infection virale chez les patients atteints de lymphadénite nécrosante histiocytaire à Taiwan. Détection du virus d’Epstein-Barr, du virus lymphotrope humain à cellules T de type I et du parvovirus B19. Am J Clin Pathol 113 : 774-781.
- Tsang WY, Chan JK, Ng CS (1994) Lymphadénite de Kikuchi. Une analyse morphologique de 75 cas avec une référence spéciale aux caractéristiques inhabituelles. Am J Surg Pathol 18 : 219-231.
- Dorfman RF, Berry GJ (1988) Kikuchi’s histiocytic necrotizing lymphadenitis : an analysis of 108 cases with emphasis on differential diagnosis. Semin Diagn Pathol 5 : 329-345.
- Aneja A, Maheswari K U1, H J GD1, Sheikh S2 (2014) Un cas rare de lymphadénopathie multifocale chez un jeune homme. Oxf Med Case Reports 2014 : 141-144.