




Solid Pseudopapillary Neoplasm of the Pancreas | Cirugía Española (English Edition)
Solid pseudopapillary tumours of the pancreas are extremely rare epithelial tumours with a limited potential for malignancy. Stanowią one mniej niż 1%-2% wszystkich zewnątrzwydzielniczych guzów trzustki.1 Nowotwory te zostały pierwotnie opisane w 1952 roku2 i od tego czasu otrzymały różne nazwy: papillary tumour of the pancreas, Frantz tumour, solid cystic papillary epithelial neoplasm, papillary cystic neoplasm, a od 1996 roku nazywane są solid pseudopapillary tumor of the pancreas.3 Najczęściej występują u młodych kobiet pochodzenia azjatyckiego lub afrykańskiego w wieku od 20 do 40 lat, chociaż zdarzały się pojedyncze przypadki u dzieci i mężczyzn.
Przedstawiamy przypadek 17-letniej pacjentki, która skarżyła się na ból brzucha w nadbrzuszu i uczucie wczesnej sytości, postępujące od kilku miesięcy, bez innych objawów. Gastroskopia wykazała ślady zewnątrzwątrobowego ucisku żołądka na trzon żołądka. W tomografii komputerowej jamy brzusznej i rezonansie magnetycznym (ryc. 1) wykryto 5 cm litą masę zaotrzewnową zależną od trzonu trzustki. Endoskopowa ultrasonografia wykazała, że była to lita zmiana hiperwaskularna w trzonie/ogonie trzustki. FNA wskazała na rozpoznanie litego pseudopapilarnego nowotworu trzustki. Wyniki badań laboratoryjnych, w tym poziomy markerów nowotworowych, mieściły się w zakresie normy.

MRI: 5cm large, well-defined, encapsulated tumor with solid-cystic component.
Wobec podejrzenia rozpoznania wykonano laparoskopową dystalną pankreatektomię z zachowaniem śledziony i naczyń śledzionowych (laparoskopowa technika Mallet-Guy),4 bez incydentu. Powrót do zdrowia po operacji przebiegał bez powikłań, a pacjent został wypisany w szóstym dniu po operacji.
Ostateczna analiza patologiczna potwierdziła rozpoznanie pseudotorbielowatego nowotworu trzustki, bez inwazji naczyniowej lub okołonaczyniowej (ryc. 2). Badanie immunohistochemiczne było silnie dodatnie dla CD56, CD10 i beta-kateniny. Progesteron i synaptofizyna wykazywały ogniskową dodatniość, a cytokeratyna AE1-AE3 i chromogranina były ujemne.
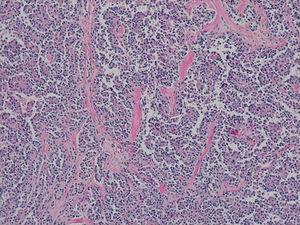
Wygląd histologiczny preparatu chirurgicznego: wzór pseudopapilarny z małymi jądrami i bez atypii (niektóre z podłużnymi szczelinami), z obecnością globulek hialinowych (hematoksylina-eozyna ×10).
Pseudopapilarne guzy trzustki są bardzo rzadkimi nowotworami trzustki o nieznanej etiologii, które dotyczą głównie młodych kobiet w drugiej i trzeciej dekadzie życia. Postuluje się, że ich pochodzenie może być nabłonkowe przewodowe, neuroendokrynne, z pluripotencjalnych komórek macierzystych, a nawet z pozatrzustkowych komórek płciowych.5 Rokowanie jest korzystne nawet w przypadku obecności przerzutów odległych, a wskaźniki przeżycia przekraczające 10 lat opisywano nawet w przypadku obecności przerzutów do wątroby lub otrzewnej.6 Objawy kliniczne są niespecyficzne i związane z wielkością guza, choć zwykle obejmują ból brzucha, uczucie pełności lub obecność masy brzusznej.7
Badania laboratoryjne są zwykle prawidłowe, a najczęstszą lokalizacją jest ogon trzustki, a następnie trzon.8 Rozpoznanie opiera się zwykle na badaniach obrazowych (USG, TK i MRI), które wykazują dobrze zarysowaną masę, która jest zamknięta i niejednorodna (lito-cystyczna) ze sporadycznymi zwapnieniami i obszarami martwiczymi.9 W diagnostyce różnicowej należy uwzględnić cystadenoma, cystadenocarcinoma, mucinous cystic neoplasms, pancreatoblastomas, teratomas i pancreatic neuroendocrine tumours jako najczęstsze zmiany hiperwaskularne. Rozpoznanie należy podejrzewać u młodych kobiet z hiperwaskularnymi lito-cystycznymi zmianami trzustki, a w przypadkach wątpliwych FNA z ultrasonografią endoskopową może potwierdzić rozpoznanie przedoperacyjne.10,11 W diagnostyce różnicowej z guzami neuroendokrynnymi, z których większość wykazuje obecność receptorów somatostatynowych, można zastosować OctreoScan®, ponieważ lite nowotwory rzekomobrodawkowe nie mają tego typu receptorów.
Ostateczne rozpoznanie ustala się na podstawie biopsji, a zalecanym leczeniem jest resekcja chirurgiczna. W 85% przypadków w momencie rozpoznania choroba jest ograniczona do trzustki, a w pozostałych przypadkach w momencie rozpoznania występują przerzuty.
Najczęstszą lokalizacją przerzutów jest wątroba, regionalne węzły chłonne, krezka, jelito grube i otrzewna.
Leczeniem z wyboru jest leczenie chirurgiczne; limfadenektomia nie jest jednak zalecana w przypadku ogniskowej postaci choroby. W przypadku obecności przerzutów lub inwazji miejscowej leczenie chirurgiczne jest nadal leczeniem z wyboru. W analizie patologicznej charakterystyczna jest obecność obszarów litych na przemian z obszarami pseudopapilarnymi, chociaż ostatnio pojawiły się doniesienia o zwiększonej ekspresji jądrowej i cytoplazmatycznej E-kadheryny i beta-kateniny jako swoistych markerów.12
Częstość występowania złośliwych litych nowotworów pseudopapilarnych lub litego raka pseudopapilarnego wynosi 15%. Niektóre cechy histologiczne są związane z agresywnym zachowaniem, takie jak wysoki wskaźnik mitotyczny, atypia jądrowa, rozległa martwica, obszary sarkomatoidalne i ekspresja Ki-67.13 Ponadto zaproponowano Ki-67 jako wskaźnik potencjału złośliwości, tak więc niski wskaźnik (poniżej 5%) wskazuje na powolny wzrost guza i lepsze rokowanie.14,15 Rola adiuwantowej radioterapii lub chemioterapii nie jest jasna, chociaż są one na ogół zarezerwowane dla przypadków nieresekcyjnych.
Niemniej jednak, chociaż resekcja chirurgiczna jest na ogół lecznicza, zalecana jest obserwacja w celu rozpoznania nawrotów miejscowych i przerzutów odległych. Całkowite 5-letnie przeżycie wynosi 95%.7,16,17
.