




Vezi articolele recente
Abstract
Introducere: Boala Kikuchi-Fujimoto, sau limfadenita necrozantă histiocitară, este o afecțiune rară cu o evoluție benignă care mimează malignitatea la prezentare. Examinarea atentă și confirmarea histologică a diagnosticului este, prin urmare, critică.
Prezentarea cazului: O femeie în vârstă de 53 de ani care s-a prezentat cu febră, limfadenopatie generalizată și simptome constituționale a fost supusă unei biopsii a ganglionilor limfatici după ce a fost îngrijorată pentru limfom malign.
Management și rezultat: Pacienta a fost diagnosticată cu boala Kikuchi-Fujimoto după confirmarea histopatologică a necrozei cu histiocite abundente și neutrofile absente.
Discuție: Febra și limfadenopatia comportă un diferențial larg, iar un diagnostic ratat al unei afecțiuni rare precum boala Kikuchi-Fujimoto poate duce la un tratament inadecvat într-o afecțiune altfel benignă.
Introducere
Boala Kikuchi-Fujimoto (KFD), sau limfadenita necrozantă histiocitară Kikuchi, este o afecțiune benignă, autolimitată, caracterizată de obicei prin febră și limfadenopatie focală. KFD are o distribuție la nivel mondial și afectează ambele sexe, cu o prevalență mai mare la femeile tinere și la persoanele asiatice . Incidența reală nu este cunoscută. Etiologia KFD este încă dezbătută în literatura de specialitate, însă au fost implicați agenți infecțioși, iar studii mai recente sugerează o etiologie virală pe baza histologiei și a clinicopatologiei asociate . Deși o asociere cu lupusul eritematos sistemic (LES) a fost dezbătută în întreaga literatură, nicio relație definitivă nu a fost dovedită. Interesant este faptul că LES trebuie încă exclus înainte de a confirma un diagnostic. KFD se poate prezenta cu simptome care durează de obicei între una și patru luni, însă recurența
a fost raportată la 3-4% dintre persoanele afectate . Prezentăm un caz de KFD la o femeie cu febră, limfadenopatie generalizată și simptome sistemice care au fost diagnosticate prin biopsie ganglionară, care a fost solicitată inițial pe baza unei suspiciuni de limfom malign.
Prezentare de caz
O femeie afro-americană în vârstă de 53 de ani s-a prezentat cu un istoric de două săptămâni de febră, transpirații nocturne, oboseală, pierdere în greutate neintenționată și limfadenopatie dureroasă la nivelul gâtului. Ea a recunoscut un istoric de șase luni de limfadenopatie la fel de dureroasă în axilă. Istoricul ei era semnificativ pentru durerile de cap, precum și pentru durerile cronice de spate auto-medicate cu marijuana. Un test de depistare a antigenului HIV din urmă cu un an a fost negativ.
Examenul fizic a evidențiat ganglioni limfatici sensibili, palpabili în lanțul cervical posterior, zona auriculară, axila și zona femurală. Cel mai mare ganglion a fost găsit în lanțul cervical posterior drept și a măsurat 1,43 cm prin ecografie (Figura 1).

Figura 1. Ganglion limfatic de 1,43 cm în lanțul cervical posterior drept pe ecografie
Rezultatele inițiale de laborator au evidențiat un număr de globule albe (WBC) de 3.000 compus din 50% limfocite, o rată de sedimentare a eritrocitelor (ESR) de 105 și o proteină C-reactivă (CRP) de 32,3. Valorile inițiale ale ESR și CRP obținute în anul anterior au fost de 10 și, respectiv, 1,6. Anticorpii ANA, lupus anticoagulant și anticorpii anti-smith au fost toți negativi. O puncție lombară a fost notabilă pentru LCR cu un WBC de 12 și proteine de 47. Antigenul EBV și IgG a fost pozitiv, precum și Varicella IgG. Culturile de LCR și frotiul acido-fast au fost negative, la fel ca și culturile de țesut, sânge și fungice. Screeningul pentru sifilis, CMV PCR și serologia pentru antigenul criptococic, coccidioide și toxoplasmoză au fost, de asemenea, negative. Tomografia computerizată cu contrast a gâtului și a toracelui a demonstrat un nodul pulmonar de 5 mm, niciun ganglion limfatic mărit și nicio dovadă de embolie pulmonară acută.
Simptomele constituționale ale pacientului au fost inițial îngrijorătoare pentru un nou diagnostic de limfom malign și a fost efectuată o biopsie nucleară ghidată ecografic și FNA a unui ganglion limfatic în triunghiul cervical posterior drept. Biopsia a demonstrat secțiuni ale ganglionului limfatic cu zone focale de necroză în interiorul cortexului caracterizate de material fibrinoid care conținea numeroase fragmente nucleare mici (figura 2). În interiorul și în jurul acestor focare se afla o populație de histiocite și celule limfoide transformate. Celulele plasmatice erau prezente în mod focal, dar neutrofilele și inflamația granulomatoasă erau absente (figura 3). Colorația imunohistochimică a evidențiat histiocite CD68 pozitive cu câteva limfocite B CD20 pozitive în zonele necrotice și multe celule mai mari, CD3 pozitive, în concordanță cu originea celulelor T .
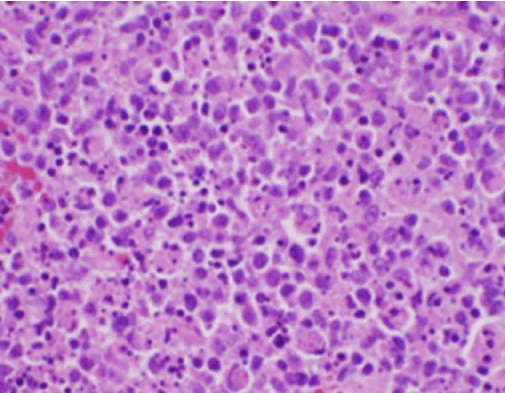
Figura 2. Colorația H&E a biopsiei ganglionului limfatic care demonstrează necroza și absența neutrofilelor
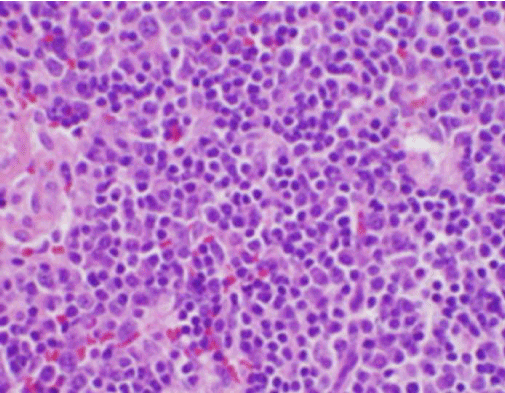
Figura 3. Colorație H&E a biopsiei ganglionului limfatic cervical care prezintă numeroase histiocite și neutrofile absente
Pacientul a fost diagnosticat cu KFD pe baza constatărilor histogologice de limfadenită necrozantă, histiocite CD68 pozitive, necroză în absența neutrofilelor și o abundență de histiocite. Restul evoluției sale spitalicești a fost fără evenimente și a fost ulterior externată sub tratament cu glucocorticoizi, cu urmărire de către reumatologie.
Discuție
Boala Kikuchi-Fujimoto, sau limfadenita necrozantă histiocitară, a fost descrisă pentru prima dată la femei tinere din Japonia în 1972 și de atunci a fost un subiect de studiu în întreaga literatură. Se caracterizează prin febră și limfadenopatie, însă pot fi prezente o varietate de semne și simptome asociate, inclusiv simptome sistemice, cum ar fi oboseala, pierderea în greutate, transpirațiile nocturne și alte manifestări mai puțin frecvente, cum ar fi meningita aseptică, splenomegalia, erupțiile cutanate și artralgiile . KFD se prezintă cel mai frecvent ca febră și limfadenopatie cervicală unilaterală la femeile tinere, însă limfadenopatia poate fi, de asemenea, multifocală. Afectarea extranodală a pielii și a măduvei osoase, de exemplu, este adesea însoțită de simptome sistemice, inclusiv transpirații nocturne, pierdere în greutate, oboseală, greață, vărsături și diaree .
Febră și limfadenopatie observate în KFD poartă un diagnostic diferențial care este destul de larg și include etiologii infecțioase, autoimune și maligne. Diagnosticul diferențial pentru febră și limfadenopatie este divers și cuprinde o gamă largă de boli cu diferite intervale de prognostic. Diagnosticarea greșită a unui pacient cu o afecțiune care are un prognostic nefavorabil și, în special, un regim de tratament mai agresiv poate duce la o îngrijire necorespunzătoare și la consecințe medicale nefaste. Mai multe rapoarte au demonstrat că simptomatologia KFD a fost confundată cu afecțiuni precum tuberculoza și limfomul malign . Ca atare, este important ca medicii să fie conștienți și să includă boala Kikuchi-Fujimoto, deși este o afecțiune rară cu o prezentare variabilă, în cadrul diagnosticului diferențial pentru febră și limfadenopatie.
Patogenia exactă rămâne incertă, însă poate semăna clinic cu o serie de afecțiuni care au un prognostic mai grav, cum ar fi tuberculoza (TB), toxoplasmoza, sifilisul, limfogranulomul veneric, LES și limfomul . Într-o serie de cazuri realizată de Fujimoto, 58% dintre pacienți au fost diagnosticați greșit și tratați în mod necorespunzător pentru TB ganglionară înainte ca histopatologia să confirme diagnosticul de KFD . Un raport a discutat un caz de KFD cu manifestări cutanate care a fost diagnosticat inițial ca un limfom cu celule mari. Pacientul a fost supus unui ciclu de terapie citotoxică înainte de a se pune diagnosticul corect .
Diagnosticul de KFD se face pe biopsia ganglionului limfatic care demonstrează focare paracorticale de necroză coagulativă cu resturi cariocrate și un infiltrat celular histiocitar . Constatarea histopatologică a resturilor nucleare fragmentate sugerează că apoptoza este mecanismul de moarte celulară, iar constatările imunohistochimice, inclusiv creșterea Fas/FasL și a celulelor T CD 8+, sprijină și mai mult acest mecanism. Constatările de laborator sunt de obicei normale, dar pot apărea ESR ridicat și leucopenie. În plus, rezultatele de laborator pot prezenta leucopenie, VSG crescută și anemie. Nu există un tratament eficient dincolo de gestionarea simptomelor cu antipiretice, analgezice,
și odihnă. Cu toate acestea, unele rapoarte au arătat că pacienții cu simptome severe pot beneficia de corticosteroizi .
Concluzie
Boala Kikuchi-Fujimoto este rară, dar conștientizarea acesteia în rândul clinicienilor este crucială atunci când se ia în considerare diagnosticul diferențial pentru un pacient care se prezintă cu simptome de febră și limfadenopatie. KFD este o afecțiune autolimitată cu un prognostic excelent, iar managementul este de obicei de susținere cu repaus și AINS pentru ameliorarea simptomelor. Atunci când este confundată cu o boală mai severă, cum ar fi tuberculoza sau limfomul malign, pacienții pot fi supuși inutil unor tratamente agresive.
- Bosch X, Guilabert A, Miquel R, Campo E (2004) Enigmatica boală Kikuchi-Fujimoto: o analiză cuprinzătoare. Am J Clin Pathol 122: 141-152.
- Chiu CF, Chow KC, Lin TY, Tsai MH, Shih CM, et al. (2000) Infecție virală la pacienții cu limfadenită histiocitară necrozantă în Taiwan. Detectarea virusului Epstein-Barr, a virusului limfotropic al celulelor T umane de tip I și a parvovirusului B19. Am J Clin Pathol 113: 774-781.
- Tsang WY, Chan JK, Ng CS (1994) Limfadenita lui Kikuchi. O analiză morfologică a 75 de cazuri cu referire specială la caracteristicile neobișnuite. Am J Surg Pathol 18: 219-231.
- Dorfman RF, Berry GJ (1988) Kikuchi’s histiocytic necrotizing lymphadenitis: an analysis of 108 cases with emphasis on differential diagnosis. Semin Diagn Pathol 5: 329-345.
- Aneja A, Maheswari K U1, H J GD1, Sheikh S2 (2014) Un caz rar de limfadenopatie multifocală la un bărbat tânăr. Oxf Med Case Reports 2014: 141-144.