




Neoplasia pseudopapilar sólida del páncreas | Cirugía Española (Spanish Edition)
Los tumores pseudopapilares sólidos del páncreas son tumores epiteliales extremadamente raros con un potencial de malignidad limitado. Representan menos del 1%-2% de todos los tumores pancreáticos exocrinos.1 Estas neoplasias fueron descritas inicialmente en 19592 y desde entonces han recibido diferentes nombres: tumor papilar del páncreas, tumor de Frantz, neoplasia epitelial papilar quística sólida o neoplasia quística papilar, y desde 1996 se denominan tumor pseudopapilar sólido del páncreas.3 Afectan con mayor frecuencia a mujeres jóvenes de ascendencia asiática o africana de entre 20 y 40 años, aunque se han dado casos aislados en niños y en hombres.
Presentamos el caso de una paciente de 17 años que se quejaba de dolor abdominal epigástrico y sensación de saciedad precoz de varios meses de evolución, sin otros síntomas. La gastroscopia mostró evidencia de compresión gástrica extrínseca en el cuerpo del estómago. La TC abdominal y la RMN (Fig. 1) detectaron una masa sólida retroperitoneal de 5 cm dependiente del cuerpo del páncreas. La ecografía endoscópica mostró que se trataba de una lesión sólida hipervascular en el cuerpo/cola del páncreas. La PAAF indicó el diagnóstico de neoplasia sólida pseudopapilar del páncreas. Los resultados de laboratorio, que incluían los niveles de marcadores tumorales, estaban dentro de los rangos normales.

RM: tumor encapsulado de 5 cm de tamaño, bien definido, con componente sólido-quístico.
Dado el diagnóstico de sospecha, se realizó pancreatectomía distal laparoscópica con preservación del bazo y los vasos esplénicos (técnica laparoscópica de Mallet-Guy),4 sin incidencias. La recuperación postoperatoria fue sin incidencias y el paciente fue dado de alta al sexto día de postoperatorio.
El análisis patológico definitivo confirmó el diagnóstico de neoplasia pseudopapilar de páncreas, sin invasión vascular ni perineural (Fig. 2). El estudio inmunohistoquímico fue fuertemente positivo para CD56, CD10 y beta-catenina. La progesterona y la sinaptofisina mostraron positividad focal, y la citoqueratina AE1-AE3 y la cromogranina fueron negativas.
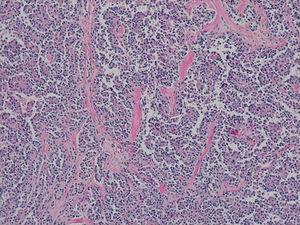
Aspecto histológico de la muestra quirúrgica: patrón pseudopapilar con núcleos pequeños y sin atipias (algunos con fisuras longitudinales), con presencia de glóbulos hialinos (hematoxilina-eosina ×10).
Los tumores pseudopapilares de páncreas son neoplasias pancreáticas muy poco frecuentes de etiología desconocida que afectan principalmente a mujeres jóvenes en la segunda y tercera décadas de la vida. Se ha propuesto que su origen podría ser ductal epitelial, neuroendocrino, de células madre pluripotentes e incluso genital extrapancreático.5 El pronóstico es favorable incluso en presencia de metástasis a distancia, y se han descrito tasas de supervivencia de más de 10 años incluso en presencia de metástasis hepáticas o peritoneales.6 Las manifestaciones clínicas son inespecíficas y se relacionan con el tamaño del tumor, aunque suelen incluir dolor abdominal, sensación de plenitud o la presencia de una masa abdominal.7
Los análisis de laboratorio suelen ser normales y la localización más frecuente es la cola del páncreas, seguida del cuerpo.8 El diagnóstico suele basarse en las pruebas de imagen (ecografía, TAC y RMN), que demuestran una masa bien delimitada, encapsulada y heterogénea (sólido-quística) con calcificaciones y áreas necróticas ocasionales.9 El diagnóstico diferencial debe hacerse con el cistadenoma, el cistadenocarcinoma, las neoplasias quísticas mucinosas, los pancreatoblastomas, los teratomas y los tumores neuroendocrinos pancreáticos como lesiones hipervasculares más frecuentes. El diagnóstico debe sospecharse en las lesiones pancreáticas sólido-quísticas hipervasculares en mujeres jóvenes y, en caso de duda, la PAAF con ecografía endoscópica puede confirmar el diagnóstico preoperatorio.10,11 Para el diagnóstico diferencial con los tumores neuroendocrinos, la mayoría de los cuales presentan receptores de somatostatina, podría utilizarse el OctreoScan® ya que las neoplasias pseudopapilares sólidas carecen de este tipo de receptores.
El diagnóstico definitivo se determina mediante biopsia, y el tratamiento recomendado es la resección quirúrgica. El 85% de los casos están limitados al páncreas en el momento del diagnóstico, y el resto han hecho metástasis en el momento del diagnóstico.
Las localizaciones más frecuentes de las metástasis son el hígado, los ganglios linfáticos regionales, el mesenterio, el epiplón y el peritoneo.
El tratamiento de elección es la cirugía; la linfadenectomía, sin embargo, no se recomienda cuando la presentación es focalizada. Cuando hay metástasis o invasión local, la cirugía sigue siendo el tratamiento de elección. En el análisis patológico, es característica la presencia de áreas sólidas alternando con áreas pseudopapilares, aunque existe un informe reciente sobre el aumento de la expresión nuclear y citoplasmática de E-cadherina y beta-catenina como marcadores específicos.12
La incidencia de neoplasias pseudopapilares sólidas malignas o carcinoma pseudopapilar sólido es del 15%. Ciertas características histológicas se han asociado a un comportamiento agresivo, como la alta tasa mitótica, la atipia nuclear, la necrosis extensa, las áreas sarcomatoides y la expresión de Ki-67.13 Además, el Ki-67 se ha propuesto como un indicador del potencial maligno, de modo que una tasa baja (inferior al 5%) indica un crecimiento tumoral lento y un mejor pronóstico.14,15 El papel de la radioterapia o la quimioterapia adyuvantes no está claro, aunque generalmente se reservan para los casos irresecables.
No obstante, aunque la resección quirúrgica es generalmente curativa, se recomienda el seguimiento para diagnosticar las recidivas locales y las metástasis a distancia. La supervivencia global a 5 años es del 95%.7,16,17