




Mécanisme – Comment l’ibuprofène (Advil ; Motrin) inhibe l’activité antithrombotique de l’aspirine
Toute personne qui effectue une simple recherche documentaire rencontrera une pléthore d’articles discutant, débattant et même argumentant sur ce sujet.Les raisons de tous ces problèmes, à notre avis, sont liées aux nombreuses incohérences entre les études concernant les doses et la durée de l’ibuprofène utilisé, le moment de l’administration de l’ibuprofène par rapport à l’administration d’aspirine, la dose et la formulation (entérique ou non entérique) de l’aspirine utilisée, la population de patients étudiée (volontaires sains vs. patients souffrant de maladies cardiovasculaires connues), l’utilisation ou non de marqueurs de laboratoire de substitution par rapport aux tests qui évaluent réellement l’agrégation plaquettaire, et enfin la conception de l’étude utilisée par les investigateurs pour générer leurs résultats.1-6 Il est donc très difficile, voire presque impossible, d’extrapoler les données actuelles de chacune de ces études, qui présentent toutes des limites ou des incohérences entre elles, et d’apporter une réponse définitive qui puisse se traduire par des critères d’évaluation cliniquement significatifs et applicables à la population générale. Certaines des divergences dans la littérature peuvent être dues à la capacité des plaquettes à s’agréger à des moments où les concentrations d’anti-inflammatoires non stéroïdiens (AINS) sont faibles, par opposition à un stade précoce après l’administration, lorsque les concentrations sont plus élevées.7 Au moment où l’ibuprofène est libéré du site de liaison de la COX-1, une partie de l’aspirine aura déjà été éliminée de l’organisme.
L’avertissement de la Food & Drug Administration (FDA) aux professionnels de la santéa récemment déclaré : « Les patients qui utilisent de l’aspirine à libération immédiate (non entérosoluble) et prennent une dose unique d’ibuprofène 400 mg doivent prendre l’ibuprofène au moins 30 minutes ou plus après l’ingestion d’aspirine, ou plus de 8 heures avant l’ingestion d’aspirine pour éviter l’atténuation de l’effet de l’aspirine ». En outre, il existe un certain nombre d’études dont les résultats sont contradictoires. « 8 Sur la base de cette recommandation, l’objectif de ce numéro n’est pas de critiquer chaque étude publiée sur ce sujet, mais plutôt d’expliquer le mécanisme proposé pour l’interaction médicamenteuse et de souligner certains des problèmes liés à son interprétation par rapport à la littérature médicale.
Que se passe-t-il lors d’une agrégation plaquettaire normale ?
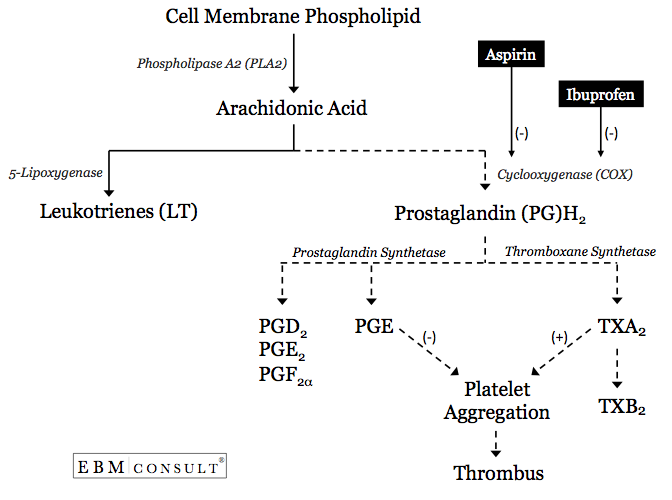
- Le processus d’augmentation de l’agrégation plaquettaire commence par la libération d’acide arachidonique (AA) de la membrane cellulaire d’une plaquette6.
- L’AA peut ensuite suivre l’une des deux voies métaboliques, la voie de la lipoxygénase, qui va générer des leucotriènes (LT), ou la voie de la cyclooxygénase (COX)-1 pour former la prostaglandine (PG) H2.9 La prostaglandine H2 peut ensuite être métabolisée par la prostaglandinesynthétase pour former d’autres PG ou elle peut être métabolisée par la thromboxane (TX)synthétase pour former le TXA2.
- Si le TXA2 est formé, l’agrégation plaquettaire sera facilitée ou encouragée.10 Cela se produit spécifiquement lorsque l’AA est capable de voyager à travers un canal hydrophobe où il peut entrer en contact avec le site catalytique au sein de l’enzyme COX-1. Si ce canal ou les zones entourant le site catalytique sont bloqués, l’AA ne pourra pas être métabolisé en PGH2 puis en TXA2, réduisant ainsi la probabilité d’agrégation plaquettaire.6,9,11
Comment l’aspirine interfère-t-elle alors avec l’agrégation plaquettaire ?
- Lorsque l’aspirine est administrée, elle acétyle de façon irréversible un résidu sérine en position 529 dans le canal hydrophobe,qui est à proximité immédiate du site catalytique où l’AA peut être métabolisé en TXA2 dérivé des plaquettes par l’enzyme COX-1.6,9,11 L’acétylation de cette zone crée un blocage où l’AA ne pourra pas accéder au site catalytique de la COX-1.
- Puisque l’aspirine fait cela de manière irréversible, la capacité de ce site catalytique au sein de l’enzyme COX-1 à métaboliser l’AA est bloquée ou inhibée pour la vie de cette plaquette (généralement autour de 7-12 jours). C’est l’une des principales raisons pour lesquelles l’aspirine confère un bénéfice cardioprotecteur contre les événements cardiovasculaires lorsqu’elle est principalement utilisée pour la prévention secondaire.
- Donc, si quelque chose d’autre concurrence ou bloque l’accès de l’aspirine à l’acétylation de ce résidu sérine au sein de l’enzyme COX-1, les bénéfices cardioprotecteurs peut-être diminués.
Comment l’ibuprofène interfère-t-il avec l’activité pharmacologique de l’aspirine ?
- Comme tous les AINS, l’ibuprofène est un inhibiteur réversible et compétitif du site catalytique du métabolisme de l’AA au sein du canal hydrophobe de l’enzyme COX-17,12.
- La présence d’ibuprofène au sein de ce canal hydrophobe bloque de manière compétitive l’accès de l’aspirine à l’acétylation du résidu sérine qui se trouve à proximité du site catalytique de l’AA.7,12,13
- Le degré d’inhibition de l’accès de l’aspirine à l’exercice de son effet pharmacologique par l’ibuprofène va être influencé par un certain nombre d’agents.
Le premier facteur et le plus évident concerne l’ordre dans lequel l’aspirine et l’ibuprofène sont administrés l’un par rapport à l’autre. Si l’aspirine est administrée en premier, elle aura accès à l’acétylation irréversible du résidu sérine au sein de l’enzyme COX-1. N’oubliez pas qu’une fois que l’aspirine a inhibé de manière irréversible l’enzyme COX-1, l’effet antiplaquettaire continuera d’exister pendant toute la durée de vie de la plaquette. Le facteur suivant est la concentration dibuprofène présente par rapport au moment de la co-administration de laspirine. L’inhibition de l’ibuprofène étant compétitive, l’agrégation plaquettaire n’est pas seulement influencée par la concentration d’ibuprofène présente, mais elle est également réversible par nature. Par conséquent, au fur et à mesure que les niveaux de médicament diminuent par les voies d’élimination, la quantité d’ibuprofène capable de bloquer l’accès de l’aspirine à son site actif diminue également, en particulier en raison de sa courte demi-vie de 2 à 4 heures.14 Cette caractéristique pharmacocinétique de l’ibuprofène est la raison pour laquelle il doit être redosé plusieurs fois au cours de la journée, alors que l’aspirine n’est dosée qu’une fois par jour. Par conséquent, on peut comprendre pourquoi il y a des variations dans les résultats des multiples études publiées dans la littérature médicale. Ainsi, l’impact clinique de cette interaction médicamenteuse estinfluencé par l’ordre dans lequel les deux médicaments sont administrés, la dose et la formulation de l’aspirine utilisée, la dose et la fréquence d’administration de l’ibuprofène utilisée, la population de patients étudiée et le type de critère d’évaluation de cette étude.
En fin de compte, la vraie question est de savoir si cette interaction se traduit par un résultat prédéfini, cliniquement pertinent, orienté vers le patient et défini sur le plan cardiovasculaire. A notre connaissance, aucun essai clinique prospectif de ce type, conçu de manière appropriée, n’a été réalisé pour répondre à cette question avec des données convaincantes dans la population de patients en question.
- GengoFM, Rubin L, Robson M et al. Effets de l’ibuprofène sur l’ampleur et la durée de l’inhibition de l’agrégation plaquettaire par l’aspirine : conséquences cliniques dans la prophylaxie des accidents vasculaires cérébraux. J Clin Pharmacol 2008;48:117-22.
- GladdingPA, Webster MWI, Farrell HB et al. L’effet antiplaquettaire de six anti-inflammatoires non stéroïdiens et leurs interactions pharmacodynamiques avec l’aspirine chez des volontaires sains. Am J Cardiol 2008;101:1060-1063.
- CryerB, Berlin RG, Copper SA et al. Double-blind, randomized, parallel,placebo-controlled study of ibuprofen effects on thromboxane B2 concentrationsin aspirin-treated healthy adult volunteers. Clin Ther2005;27:185-191.
- MacDonaldTM, Wei L et al. Effect of ibuprofen on cardioprotective effect ofaspirin. Lancet 2003;361:573-74.
- KurthT, Glynn RJ, Walker AM et al. Inhibition des avantages cliniques de l’aspirine sur le premier infarctus du myocarde par les anti-inflammatoires non stéroïdiens. Circulation 2003;108:1191-1195.
- Catella-LawsonF, Reilly MP, Kapoor SC et al. Les inhibiteurs de cyclooxygénase et les effets antiplaquettaires de l’aspirine. N Engl J Med 2001;345:1809-17.
- EvansAM. Pharmacodynamique et pharmacocinétique des profens:énantiosélectivité, implications cliniques, et référence spéciale auS(+)-ibuprofène. J Clin Pharmacol 1996;36:7S-15S.
- Food& Drug Administration. Information pour les professionnels de santé:utilisation concomitante d’ibuprofène et d’aspirine. Département américain de la santé &Services à la personne. Dernier accès : 09-19-2011.
- FunkCD, Funk LB, Kennedy ME et al. Human platelet/erythroleukemia cellprostaglandin G/H synthase : cDNA cloning, expression, and gene chromosomalassment. FASEB J 1991;5:2304-12.
- FitzgeraldGA. Mécanismes d’activation des plaquettes : le thromboxane A2 comme signal d’amplification d’autres agonistes. Am J Cardiol 1991;68:11B-15B.
- LollPJ, Picot D, Garavito RM. The structural basis of aspirin activityinferred from the crystal structure of inactivated prostaglandin H2synthase. Nat Struct Biol 1995;2:637-43.
- LollPJ, Picot D, Ekabo O et al. Synthèse et utilisation d’analogues iodés de médicaments anti-inflammatoires non stéroïdiens comme sondes cristallographiques du site actif de la prostaglandineH2 synthase cyclooxygénase. Biochimie 1996;35:7330-40.
- RaoGH, Johnson GG, Reddy KR et al. L’ibuprofène protège la cyclo-oxygénase plaquettaire de l’inhibition irréversible par l’aspirine. Arteriosclerosis 1983;3:383-8.