




Se de senaste artiklarna
Abstract
Introduktion: Kikuchi-Fujimotos sjukdom, eller histiocytär nekrotiserande lymfadenit, är ett sällsynt tillstånd med ett godartat förlopp som vid presentationen efterliknar malignitet. Noggrann undersökning och histologisk bekräftelse av diagnosen är därför avgörande.
Fallpresentation: En 53-årig kvinna som presenterades med feber, generaliserad lymfadenopati och konstitutionella symtom genomgick en lymfkörtelbiopsi efter oro för malignt lymfom.
Hantering och resultat: Patienten diagnostiserades med Kikuchi-Fujimotos sjukdom efter histopatologisk bekräftelse av nekros med rikliga histiocyter och frånvarande neutrofiler.
Diskussion: Patienten fick en diagnos av Kikuchi-Fujimotos sjukdom efter histopatologisk bekräftelse av nekros med rikliga histiocyter och frånvarande neutrofiler: Feber och lymfadenopati medför en bred differentialdiagnos, och en missad diagnos av ett sällsynt tillstånd som Kikuchi-Fujimotos sjukdom kan leda till olämplig behandling av ett annars godartat tillstånd.
Introduktion
Kikuchi-Fujimotos sjukdom (KFD) eller Kikuchi histiocytär nekrotiserande lymfadenit är ett godartat, självbegränsat tillstånd som typiskt kännetecknas av feber och fokal lymfadenopati. KFD är spridd över hela världen och drabbar båda könen med en högre prevalens hos unga kvinnor och asiatiska personer . Den verkliga incidensen är inte känd. Etiologin för KFD är fortfarande omdiskuterad i litteraturen, men infektiösa ämnen har varit inblandade, och nyare studier tyder på en viral etiologi på grundval av den associerade histologin och klinikopatologin . Även om ett samband med systemisk lupus erythematosus (SLE) har diskuterats i litteraturen har inget definitivt samband kunnat påvisas. Intressant nog måste SLE fortfarande uteslutas innan en diagnos bekräftas. KFD kan presentera symtom som vanligtvis varar en till fyra månader, men återfall
har rapporterats hos 3-4 % av de drabbade individerna . Vi presenterar ett fall av KFD hos en kvinna med feber, generaliserad lymfadenopati och systemiska symtom som diagnostiserades genom lymfkörtelbiopsi, som ursprungligen beställdes baserat på misstanke om malignt lymfom.
Fallpresentation
En 53-årig afroamerikansk kvinna presenterade sig med en tvåveckorshistoria av feber, nattsvettningar, trötthet, oavsiktlig viktnedgång och smärtsam lymfadenopati i halsen. Hon hade i sex månader haft en liknande smärtsam lymfadenopati i axillan. I hennes anamnes var det vanligt med huvudvärk och kronisk ryggsmärta som hon självmedicinerade med marijuana. En hiv-antigenscreening från ett år tidigare var negativ.
Fysisk undersökning avslöjade ömma, palpabla lymfkörtlar i den bakre cervikala kedjan, aurikulära området, axillan och femorala området. Den största lymfkörteln fanns i den högra bakre cervikala kedjan och mätte 1,43 cm vid ultraljud (figur 1).

Figur 1. 1,43 cm lymfkörtel i höger bakre halskedja på ultraljud
Initiala laboratorieresultat visade ett antal vita blodkroppar (WBC) på 3 000 som bestod av 50 % lymfocyter, en erytrocytsedimentationshastighet (ESR) på 105 och C-reaktivt protein (CRP) på 32,3. Baseline ESR och CRP från föregående år var 10 respektive 1,6. ANA, lupus antikoagulantia och anti-smith-antikroppar var alla negativa. En lumbalpunktion var anmärkningsvärd för CSF med en WBC på 12 och protein på 47. EBV-antigen och IgG var positivt liksom varicella IgG. CSF-odlingar och syrafasta smear var negativa, liksom vävnads-, blod- och svampodlingar. Syfilisscreening, CMV-PCR och serologi för kryptokockantigen, koccidioider och toxoplasmos var också negativa. Kontrast-CT av hals och bröst visade en 5 mm stor lungknöl, inga förstorade lymfkörtlar och inga tecken på akut lungemboli.
Patientens konstitutionella symtom var initialt oroande för en ny diagnos av malignt lymfom, och en ultraljudsstyrd kärnbiopsi och FNA av en lymfkörtel i den högra bakre cervikala triangeln utfördes. Biopsin visade sektioner av lymfkörteln med fokala områden av nekros i cortex som kännetecknades av fibrinoid material som innehöll många små kärnfragment (figur 2). Inom och runt dessa fyndpunkter fanns en population av histiocyter och transformerade lymfoida celler. Plasmaceller var fokuserat närvarande, men neutrofiler och granulomatös inflammation saknades (figur 3). Immunohistokemisk färgning visade CD68-positiva histiocyter med ett fåtal CD20-positiva B-lymfocyter i de nekrotiska områdena och många större, CD3-positiva celler som överensstämde med T-cellernas ursprung .
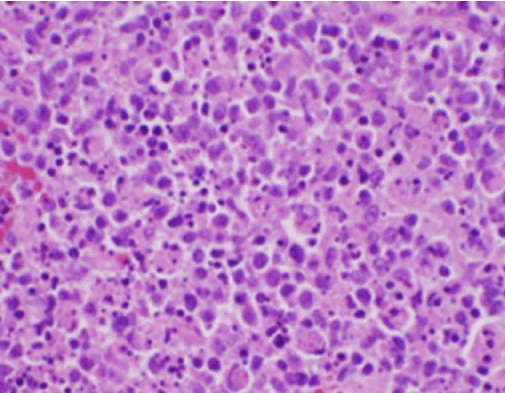
Figur 2. H&E-färgning av lymfkörtelbiopsi som visar nekros och avsaknad av neutrofiler
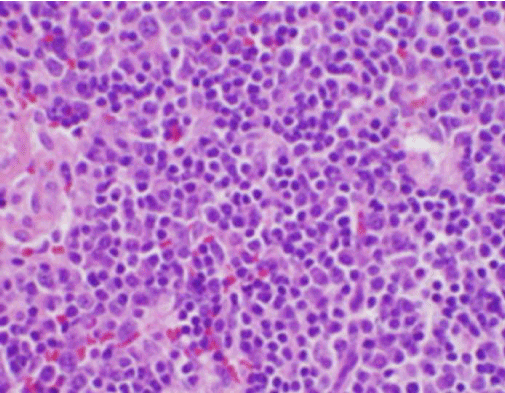
Figur 3. H&E-färgning av biopsi av cervikal lymfkörtel som visar många histiocyter och frånvarande neutrofiler
Patienten diagnostiserades med KFD baserat på de histogologiska fynden av nekrotiserande lymfadenit, CD68-positiva histiocyter, nekros i avsaknad av neutrofiler och ett överflöd av histiocyter. Resten av sjukhusförloppet var händelselöst och hon skrevs därefter ut på glukokortikoidbehandling med uppföljning av reumatologin.
Diskussion
Kikuchi-Fujimotos sjukdom, eller histiocytär nekrotiserande lymfadenit, beskrevs för första gången hos unga kvinnor i Japan 1972 och har sedan dess varit föremål för studier i hela litteraturen. Den kännetecknas av feber och lymfadenopati, men en mängd olika associerade tecken och symtom kan förekomma, inklusive systemiska symtom som trötthet, viktnedgång, nattsvettningar och andra mindre vanliga manifestationer som aseptisk hjärnhinneinflammation, splenomegali, utslag och arthralgi . KFD visar sig oftast som feber och unilateral cervikal lymfadenopati hos unga kvinnor, men lymfadenopatin kan också vara multifokal. Extranodal involvering av till exempel hud och benmärg åtföljs ofta av systemiska symtom, inklusive nattliga svettningar, viktminskning, trötthet, illamående, kräkningar och diarré .
Den feber och lymfadenopati som ses vid KFD medför en differentialdiagnos som är ganska bred och innefattar infektiösa, autoimmuna och maligna etiologier. Differentialdiagnosen för feber och lymfadenopati är mångsidig och omfattar ett brett spektrum av sjukdomar med varierande prognoser. Att feldiagnostisera en patient med ett tillstånd som har en ogynnsam prognos och i synnerhet en mer aggressiv behandlingsregim kan leda till olämplig vård och dåliga medicinska konsekvenser. Flera rapporter har visat att symptomatologin vid KFD har förväxlats med tillstånd som tuberkulos och maligna lymfom . Det är därför viktigt att kliniker är medvetna om och inkluderar Kikuchi-Fujimotos sjukdom, även om det är ett sällsynt tillstånd med en varierande presentation, i differentialdiagnosen för feber och lymfadenopati.
Den exakta patogenesen förblir osäker, men den kan kliniskt likna ett antal tillstånd med allvarligare prognoser, såsom tuberkulos (TB), toxoplasmos, syfilis, lymfogranuloma venereum, SLE och lymfom . I en fallserie av Fujimoto feldiagnostiserades och behandlades 58 % av patienterna felaktigt för tuberkulos i lymfkörteln innan histopatologin bekräftade diagnosen KFD . I en rapport diskuterades ett fall av KFD med kutana manifestationer som ursprungligen diagnostiserades som ett storcelligt lymfom. Patienten genomgick en cytotoxisk behandling innan den korrekta diagnosen ställdes .
Diagnosen av KFD ställs vid biopsi av lymfkörteln som visar parakortikala fynd av koagulativ nekros med karyorektiska rester och ett histiocytärt cellinfiltrat . Det histopatologiska fyndet av fragmenterade kärnrester tyder på apoptos som celldödsmekanism, och immunohistokemiska fynd, inklusive ökad mängd Fas/FasL och CD 8+ T-celler, stöder ytterligare denna mekanism . Laboratoriefynden är vanligtvis normala, men förhöjd ESR och leukopeni kan förekomma. Dessutom kan laboratoriefynden visa leukopeni, förhöjd ESR och anemi. Det finns ingen effektiv behandling utöver symtombehandling med febernedsättande medel, smärtstillande medel
och vila. Vissa rapporter har dock visat att patienter med svåra symtom kan ha nytta av kortikosteroider.
Slutsats
Kikuchi-Fujimotos sjukdom är sällsynt, men medvetenhet om den bland kliniker är avgörande när man överväger differentialdiagnosen för en patient som presenterar symtom på feber och lymfadenopati. KFD är ett självbegränsat tillstånd med utmärkt prognos, och behandlingen är vanligtvis stödjande med vila och NSAID-preparat för att lindra symtomen. När det misstas för en allvarligare sjukdom, till exempel tuberkulos eller malignt lymfom, kan patienterna i onödan utsättas för aggressiva behandlingar.
- Bosch X, Guilabert A, Miquel R, Campo E (2004) Enigmatic Kikuchi-Fujimoto disease: a comprehensive review. Am J Clin Pathol 122: 141-152.
- Chiu CF, Chow KC, Lin TY, Tsai MH, Shih CM, et al. (2000) Virusinfektion hos patienter med histiocytär nekrotiserande lymfadenit i Taiwan. Påvisande av Epstein-Barr-virus, typ I humant T-cells lymphotropiskt virus och parvovirus B19. Am J Clin Pathol 113: 774-781.
- Tsang WY, Chan JK, Ng CS (1994) Kikuchis lymfadenit. En morfologisk analys av 75 fall med särskild hänvisning till ovanliga drag. Am J Surg Pathol 18: 219-231.
- Dorfman RF, Berry GJ (1988) Kikuchis histiocytära nekrotiserande lymfadenit: en analys av 108 fall med tonvikt på differentialdiagnostik. Semin Diagn Pathol 5: 329-345.
- Aneja A, Maheswari K U1, H J GD1, Sheikh S2 (2014) Ett sällsynt fall av multifokal lymfadenopati hos en ung man. Oxf Med Case Reports 2014: 141-144.